Single-cellRNA-seq analysis is a rapidly evolving field at the forefront of transcriptomic research, used in high-throughput developmental studies and rare transcript studies to examine cell heterogeneity within a populations of cells.
The cellular resolution and genome wide scope make it possible to draw new conclusions that are not otherwise possible with bulk RNA-seq. The analysis requires a great deal of knowledge about statistics, wet-lab protocols, and some machine learning due to variability and sparseness of the data. The uncertainty from the low coverage and low cell numbers per sample that once were common setbacks in the field are overcome by 10x Genomics which provides high-throughput solutions which are quickly championing the field.
Era of 10x Genomics
10x genomics has provided not only a cost-effective high-throughput solution to understanding sample heterogeneity at the individual cell level, but has defined the standards of the field that many downstream analysis packages are now scrambling to accommodate.
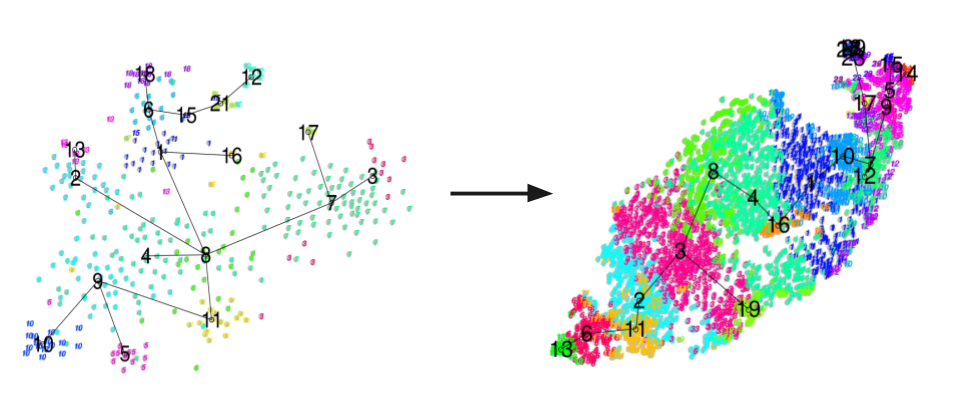
Figure 1: From less than 1K to over 10K with 10x genomics: Analyses of two separate scRNA datasets using the (left) CelSEQ2 protocol, and the (right) 10x Chromium system.
The gain in resolution reduces the granularity and noise issues that plagued the field of scRNA-seq not long ago, where now individual clusters are much easier to decipher due to the added stability added by this gain in information.
Library Preparation
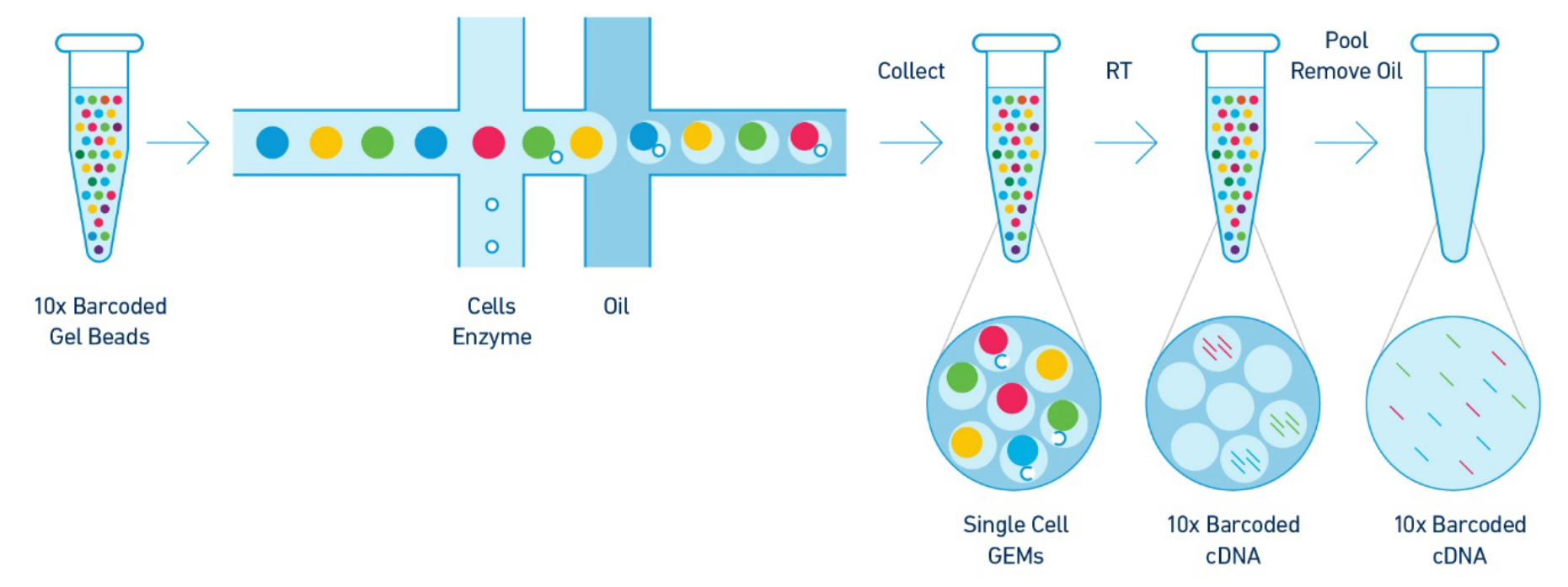
Figure 2: An overview of the library preparation
The 10X barcoded gel beads consist of a pool barcodes which are used to separately index each cell’s transcriptome. The individual gel barcodes are delivered to each cell via flow-cytometry, where each cell is fed single-file along a liquid tube and tagged with a 10X gel bead. The cells are then isolated from one another within thousands of nanoliter droplets, where each droplet described by a unique 10x barcode that all reads in that droplet are associated with once they undergo reverse-transcription (RT) which reconstructs the mRNA into a cDNA counterpart. The oil is then removed and all (now barcoded) cDNAreads are pooled together to be sequenced.
Though there are approximately 3 million 10x gel barcodes used, the amount actually qualitatively profiled in a sample is ~10,000 due to majority of droplets (>90%) being empty in order to ensure that the remainder contains only one cell.
There are actually two sets of barcodes for the different chemistries provided; one which has 737,000 barcodes, and one with ~3,7 million barcodes.
Both are provided in the Zenodo link, but we will only work with the 3 million barcodes because this is what is provided with the chemistry version.
If you are getting a low number of cells detected for your reads later on, it could be because the barcodes provided are wrong.
The first part of this tutorial is essentially a one-click “fire and forget” solution to demultiplexing and quantifying scRNA-seq data, where much of the complexity required in this extremely crucial stage is simplified into a single step.
However, those who are more interested in learning the intricacies of how FASTQ files are transformed into a count matrix, please see the Pre-processing of Single-CellRNA Data tutorial.
10x Genomics has its own processing pipeline, Cell Ranger to process the scRNA-seq outputs it produces, but this process requires much configuration to run and is significantly slower than other mappers.
Since STARsolo is a drop-in solution to the Cell Ranger pipeline, the first part of the tutorial is a one-click solution where users are encouraged to launch their RNA STARsolo jobs and spend the time familiarising themselves with the pre-processing training materials mentioned above.
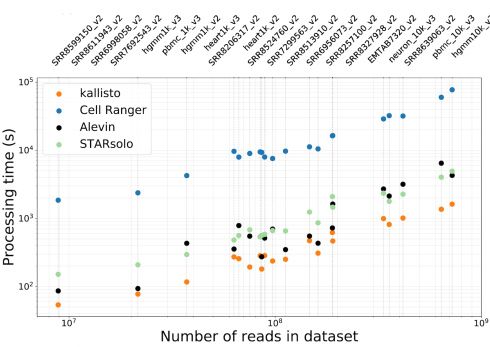
Figure 4: Benchmark of different mapping software.
The image is from “Melsted et al (June, 2019). Notice the order of magnitude speed up that STARsolo and a few others display, for a variety of different datasets in comparison to Cell Ranger.
The second part of this tutorial also has a one-click solution to producing a matrix identical to that given by the Cell Ranger pipeline, but the more interesting aspects of the pipeline are explored in the Introspective Method part of the tutorial.
Producing a Count Matrix from FASTQ
Here we will use the 1k PBMCs from a Healthy Donor (v3 chemistry) from 10x genomics, consisting of 1000 Peripheral blood mononuclear cells (PBMCs) extracted from a healthy donor, where PBMCs are primary cells with relatively small amounts of RNA (~1pg RNA/cell).
The source material consists of 6 FASTQ files split into two sequencing lanes L001 and L002, each with three reads of R1 (barcodes), R2 (cDNA sequences), I1 (illumina lane info):
pbmc_1k_v3_S1_L001_R1_001.fastq.gz
pbmc_1k_v3_S1_L001_R2_001.fastq.gz
pbmc_1k_v3_S1_L001_I1_001.fastq.gz
pbmc_1k_v3_S1_L002_R1_001.fastq.gz
pbmc_1k_v3_S1_L002_R2_001.fastq.gz
pbmc_1k_v3_S1_L002_I1_001.fastq.gz
The Cell Ranger pipeline requires all three files to perform the demultiplexing and quantification, but RNA STARsolo does not require the I1 lane file to perform the analysis. These source files are provided in the Zenodo data repository, but they require approximately 2 hours to process. For this tutorial, we will use datasets sub-sampled from the source files to contain approximately 300 cells instead of 1000. Details of this sub-sampling process can be viewed at the Zenodo link.
Data upload and organization
For the mapping, we require the sub-sampled source files, as well as a “whitelist” of (~3,7 million) known cell barcodes, freely extracted from the Cell Ranger pipeline. This whitelist file may be found within the Galaxy Data Library, but it is included here in the Zenodo record for convenience and also because the sequencing facility may not always provide this file.
The barcodes in the R1 FASTQ data are checked against these known cell barcodes in order assign a specific read to a specific known cell. The barcodes are designed in such a manner that there is virtually no chance that they will align to a place in the reference genome. In this tutorial we will be using hg19 (GRCh37) version of the human genome, and will therefore also need to use a hg19 GTF file to annotate our reads.
Hands-on: Data upload and organization
Create a new history and rename it (e.g. scRNA-seq 10X dataset tutorial)
Click the new-history icon at the top of the history panel.
If the new-history is missing:
Click on the galaxy-gear icon (History options) on the top of the history panel
Select the option Create New from the menu
Import the sub-sampled FASTQ data from Zenodo or from the data library (ask your instructor)
As an alternative to uploading the data from a URL or your computer, the files may also have been made available from a shared data library:
Go into Shared data (top panel) then Data libraries
Navigate to the correct folder as indicated by your instructor
Select the desired files
Click on the To History button near the top and select as Datasets from the dropdown menu
In the pop-up window, select the history you want to import the files to (or create a new one)
There are two main reagent kits used during the library preparation, and the choice of one will influence the size of the sequences we work with. Below we can see the layout of the primers used in both chemistries. We can ignore most of these as they are not relevant, namely: the P5 and P7 illumina primers are used in the illumina bridge amplification process; the Sample Index is an 8bp primer which is related to the Chromium system that balances nucleotide bias and ensures that there is no sample overlap during the multiplexed sequencing; and the Poly(dT) VN primer used to capture RNA sequences with poly-A tails (i.e. mRNA).
Figure 5: 10x Chromiumv2 and Chromiumv3 Chemistries
The primers of interest to us are the Cell Barcode (CB) and the Unique Molecular Identifiers (UMI) used in the Read 1 sequencing primer, as they describe to us how to demultiplex and deduplicate our reads. It is highly advised that the Plates, Batches, and Barcodes slides are revisited to refresh your mind on these concepts.
Chemistry
Read 2
Read 1 (CB + UMI)
Insert (Read 2 + Read 1)
v2
98
26 (16 + 10)
124
v3
91
28 (16 + 12)
119
The table above gives a summary of the primers used in the image and the number of basepairs occupied by each.
Unstranded protocols do not distinguish between whether a fragment was sequenced from the forward or the reverse strand, which can lead to some ambiguity if the fragment overlaps two transcripts. In the image below, it is not clear whether the fragment is derived from GeneF or GeneR due to this overlap.
Figure 6:Mapping fragments to overlapping transcripts is ambiguous with unstranded protocols.
Stranded protocols overcome this by fitting different 5’ and 3’ primers and adaptors, meaning that the orientation of the fragment is fixed during sequencing. For the Chromium v2 and v3 chemistries, the barcode information is purely within the R1 forward strand.
Question
What has stayed constant between the chemistry versions?
What advantage does this constant factor give?
What do UMIs do?
What advantage does the 2 extra bp in the v3 UMIs have over v2 UMIs?
What will be the strandedness of the generated library
The Cell Barcode (CB) has remained at 16bp for both chemistries.
This has the advantage that the same set of barcodes can be used in both chemistries, which is important because barcodes are very hard to design.
They need to be designed in such a way to minimise accidentally aligning to the reference they were prepared to be used for.
Longer barcodes tend to be more unique, so this is a problem that is being solved as the barcodes increase in size, allowing for barcodes that can be used on more than one reference to be more common, as seen above.
UMIs (or Unique Molecular identifiers) do not delineate cells as Cell Barcodes do, but instead serve as random ‘salt’ that tag molecules randomly and are used to mitigate amplification bias by deduplicating any two reads that map to the same position with the same UMI, where the chance of this happening will be astronomically small unless one read is a direct amplicon of the other.
\(4^{10} = 1,048,576\) unique molecules tagged, vs. \(4^{12} = 16,777,216\) unique molecules tagged. The reality is much much smaller due to edit distances being used that would reduce both these numbers substantially (as seen in the Plates, Batches, and Barcodes slides), but the scale factor of 16 times more molecules (\(4^{12-10} = 16\)) can be uniquely tagged is true.
Forward.
The differences in the chemistries is a slight change in the library size, where the v2 aims to capture on average 50,000 reads per cell, whereas the v3 aims to capture at minimum 20,000 reads per cell. This greatly reduces the lower-tail of the library size compared to the previous version.
Determining what Chemistry our Data Contains
To perform the demultiplexing, we need to tell RNA STARsolo where to look in the R1 FASTQ to find the cell barcodes. We can do this by simply counting the number of basepairs in any read of the R1 files.
Question
Peek at one of the R1 FASTQ files using the galaxy-eye symbol below the dataset name.
How many basepairs are there in any given read?
Which library preparation chemistry version was this read generated from?
There are 28 basepairs.
The v2 has 26 basepairs, but the v3 has 28 basepairs. Therefore the reads we have here use the Chromium v3 chemistry.
Performing the Demultiplexing and Quantification
We will now proceed to demultiplex, map, and quantify both sets of reads using the correct chemistry discovered in the previous sub-section.
Comment
RNA STARsoloTool: toolshed.g2.bx.psu.edu/repos/iuc/rna_starsolo/rna_starsolo/2.7.8a consumes a large amount of memory. During the Smörgåsbord training please use Human (Homo Sapiens): hg19 chrX as the reference genome if you follow this tutorial on usegalaxy.org. This performs the mapping only against chromosome X. The full output dataset is available at zenodo and will be the starting point for the next tutorial.
Hands-on: Hands-on
RNA STARsoloTool: toolshed.g2.bx.psu.edu/repos/iuc/rna_starsolo/rna_starsolo/2.7.8a with the following parameters:
“Custom or built-in reference genome”: Use a built-in index
“Reference genome with or without an annotation”: use genome reference without builtin gene-model
“Select reference genome”: Human (Homo Sapiens): hg19 Full or Human (Homo Sapiens): hg19 chrX
“Gene model (gff3,gtf) file for splice junctions”: Homo_sapiens.GRCh37.75.gtf
“Length of genomic sequence around annotated junctions”: 100
“Type of single-cellRNA-seq”: Drop-seq or 10X Chromium
“Input Type”: Separate barcode and cDNAreads
param-file“RNA-Seq FASTQ/FASTA file, Barcode reads”: Multi-select L001_R1_001 and L002_R1_001 using the Ctrl key.
param-file“RNA-Seq FASTQ/FASTA file, cDNAreads”: Multi-select L001_R2_001 and L002_R2_001 using the Ctrl key.
“Matching the Cell Barcodes to the WhiteList”: Multiple matches (CellRanger 2)
Under “Advanced Settings”:
“Strandedness of Library”: Forward
“Collect UMI counts for these genomic features”: Gene: Count reads matching the Gene Transcript
“Type of UMI filtering”: Remove UMIs with N and homopolymers (similar to CellRanger 2.2.0)
“Cell filter type and parameters”: Do not filter
“Field 3 in the Genes output”: Gene Expression
Comment
The in-built Cell filtering is a relatively new feature that emulates the CellRanger pipeline. Here, we set the filtering options to not filter because we will use our own methods to better understand how this works. For your own future datasets, you may wish to enable the filtering parameter.
Inspecting the Output Files
At this stage RNA STARsolo has output 6 files; a program log, a mapping quality file, a BAM file of alignments, and 3 count matrix files:
Log
Feature Statistic Summaries
Alignments
Matrix Gene Counts
Barcodes
Genes
The log and the summaries files give program logistics and metrics about the quality of the mapping. The BAM file contains the reads mapped to the reference genome. The matrix gene counts is the count matrix in matrixmarket format, accompanied by the list of genes and cell barcodes in separate files. These can be converted into tabular or AnnData formats using the toolImport Anndata and loom tool.
Mapping Quality
Let us investigate the output log. This type of quality control is essential in any RNA-based analysis and it is strongly recommended that you familiarise yourself with the Quality Control tutorial.
Hands-on: Hands-on
MultiQCTool: toolshed.g2.bx.psu.edu/repos/iuc/multiqc/multiqc/1.9+galaxy1 with the following parameters:
In “Results”:
In “1:Results”:
“Which tool was used generate logs?”: STAR
In “STAR output”:
In “1:STAR output”:
“Type of STAR output?”: Log
param-file“STAR log output”: RNA STARsolo: log
Question
What percentage of reads are uniquely mapped?
87.5%
This is good, and is expected of 10x datasets.
Quantification Quality
The above tool provides a nice visualisation of the output log, and is a fantastic way to combine multiple quality sources into one concise report.
However, sometimes it is often more informative to look directly at the source quality control file. For example, let us investigate the STARsolo Feature summaries file.
We can look at this directly by clicking on the galaxy-eye symbol of the Feature Statistic Summaries file.
The explanation of these parameters can be seen in the RNA STAR Manual under the STARsolo section, with most of the information given in the details box below.
Parameter
Explanation
nNinCB
number of reads with more than 2 Ns in cell barcode (CB)
nUMIhomopolymer
number of reads with homopolymer in CB
nTooMany
(not used at the moment)
nNoMatch
number of reads with CBs that do not match whitelist even with one mismatch
All of the above reads are discarded from Solo output.
The remaining reads are checked for any overlap with the features/genes:
Parameter
Explanation
nUnmapped
number of reads unmapped to the genome
nNoFeature
number of reads that map to the genome but do not belong to a feature
nAmbigFeature
number of reads that belong to more than one feature
nAmbigFeatureMultimap
number of reads that belong to more than one feature and are also multimapping to the genome (this is a subset of the nAmbigFeature)
nTooMany
number of reads with ambiguous CB (i.e. CB matches whitelist with one mismatch but with posterior probability 0.95)
nNoExactMatch
number of reads with CB that matches a whitelist barcode with 1 mismatch, but this whitelist barcode does not get any other reads with exact matches of CB
These metrics can be grouped into more broad categories:
nNinCB + nUMIhomopolymer + nNoMatch + nTooMany + nNoExactMatch = number of reads with CBs that do not match whitelist.
nUnmapped + nAmbigFeature = number of reads without defined feature (gene).
nMatch = number of reads that are output as solo counts.
The three categories above summed together should be equal to the total number of reads (which is also given in the MultiQC output).
The main information to gather at this stage is that the nCellBarcodes tell us how many cells were detected in our sample, where we see that there are 6101 cells. Another metric to take into account is that the number of matches (nMatch) has the largest value, and that the number of reads that map to the genome but not to a feature/gene given in the GTF (nNoFeature) is not too large. The number of no features is also quite high when mapping the original (non-subsampled) 10x input datasets, so this appears to be the default expected behaviour.
Producing a Quality Count Matrix
The matrix files produced by RNA STARsolo are in the bundled format, meaning that the information to create a tabular matrix of Genes vs Cells are separated into different files. These files are already 10x analysis datasets, compatible with any downstreamsingle-cellRNA analysis pipeline, however the number of cells represented here are greatly over-represented, as they have not yet been filtered for high quality cells, and therefore the matrix represents any cells that were unambiguously detected in the sample.
If we were to construct a cell matrix using the data we have, we would have a large matrix of 60,000 Genes against 3 million Cells, of which most values would be zero, i.e. an extremely sparse matrix.
To get a high quality count matrix we must apply the DropletUtils tool, which will produce a filtered dataset that is more representative of the Cell Ranger pipeline.
Cell Ranger Method
Hands-on: Default Method
DropletUtilsTool: toolshed.g2.bx.psu.edu/repos/iuc/dropletutils/dropletutils/1.10.0+galaxy1 with the following parameters:
“Format for the input matrix”: Bundled (barcodes.tsv, genes.tsv, matrix.mtx)
param-file“Count Data”: Matrix Gene Counts (output of RNA STARsolotool)
param-file“Genes List”: Genes (output of RNA STARsolotool)
param-file“Barcodes List”: Barcodes (output of RNA STARsolotool)
“Operation”: Filter for Barcodes
“Method”: DefaultDrops
“Expected Number of Cells”: 3000
“Upper Quantile”: 0.99
“Lower Proportion”: 0.1
“Format for output matrices”: Tabular
Comment: Default Parameter
The “Expected Number of Cells” parameter is the number of cells you expect to see in your sample, but does not correspond to how many cells you expect to see in a sub-sampled dataset like this one. The default is 3000 for Cell Ranger, which will yield ~300 cells here, but users are encouraged to experiment with this value when dealing with their own data, to recover the desired number of cells.
Question
How many cells were detected?
Does this agree with the STARsolo Feature Statistics output?
By clicking on the title of the output dataset in the history we can expand the box to see the output says that there are 272 cells in the output table.
If we expand the galaxy-eyeRNA STARsolo Feature Statistic Summaries Dataset and look at the nCellBarcodes value, we see that RNA STARsolo detected 6101 cells. What this means is that 6101 barcodes were detected in total, but only 272 of them were above an acceptable threshold of quality, based on the default upper quantile and lower proportion parameters given in the tool. Later on, we will actually later visualise these thresholds ourselves by “ranking” the barcodes, to see the dividing line between high and low quality barcodes.
This will produce a count matrix in a human readable tabular format which can be used in downstream analysis tools, such as those used in the DownstreamSingle-cellRNA analysis with RaceID tutorial.
The tabular outputs here are selected mostly for transparency, since they are easy to inspect. However, the datasets produced with 10x data are very large and very sparse, meaning there is much data redundancy due to the repetition of zeroes everywhere in the data.
Many analysis packages have attempted to solve this problem by inventing their own standard, which has led to the proliferation of many different “standards” in the scRNA-seq package ecosystem, most of which require an R programming environment to inspect.
As expected, the format that usually wins is the one which is most common in the field. In this case, the format also happens to be a very good format that stores data in a concise, compressed, and extremely readable manner:
<img src=”/training-material/topics/transcriptomics/images/tenx_anndata.svg “AnnData is an HDF5-based format, which stores gene and cell information in their own matrices, complementary to the main data matrix.” “ alt=”anddata. “ loading=”lazy”>
The AnnData format (hda5) is an extension of the HDF5 format, which supports multidimensional datasets to be stored in a consistent and space-optimised way. This is the default output of Cell Ranger and so is also the default output of RNA STARsolo. The format is also now a widely accepted format in many downstream analysis suites.
Introspective Method
The DefaultDrops method given in the previous sub-section is a good one-click solution to emulating the Cell Ranger process of producing a high quality count matrix.
However, the DropletUtils tool does provide other options for determining which cells are of “good” quality and which are not.
A useful diagnostic for droplet-based data is the barcode rank plot, which shows the (log-)total UMI count for each barcode on the y-axis and the (log-)rank on the x-axis. This is effectively a transposed empirical cumulative density plot with log-transformed axes. It is useful as it allows users to examine the distribution of total counts across barcodes, focusing on those with the largest counts.
Hands-on: Rank Barcodes
DropletUtilsTool: toolshed.g2.bx.psu.edu/repos/iuc/dropletutils/dropletutils/1.10.0+galaxy1 with the following parameters:
“Format for the input matrix”: Bundled (barcodes.tsv, genes.tsv, matrix.mtx)
param-file“Count Data”: Matrix Gene Counts (output of RNA STARsolotool)
param-file“Genes List”: Genes (output of RNA STARsolotool)
param-file“Barcodes List”: Barcodes (output of RNA STARsolotool)
“Operation”: Rank Barcodes
“Lower Bound”: 100
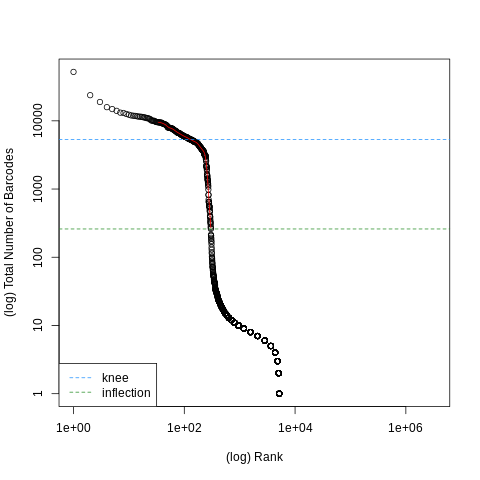
Figure 7: Barcode Ranks: The separating thresholds of high and low quality cells
The knee and inflection points on the curve mark the transition between two components of the total count distribution. This is assumed to represent the difference between empty droplets with little RNA and cell-containing droplets with much more RNA, and gives us a rough idea of how many cells to expect in our sample.
Question question Questions
How many cells do we expect to see in our sample based on the above plot?
How many high quality cells do we expect to see in our sample bed on the above plot?
What is the minimum number of UMIs that we can expect to see for high quality cells?
We see the blue knee line cross the threshold of barcodes at just below than the 10000 Rank on the horizontal log scale, which is shown in the expanded view of our data as "knee = 5300". This is in good accordance with the 6101 cells shown in the STARsolo log output previously.
This threshold is given by the inflection line, which is given at "inflection = 260", so 260 cells.
The vertical drop in the chart occurs at a log X-axis position just above 1e+02, so we can estimate ~ 200 UMIs minimum per cell.
On large 10x datasets we can use these thresholds as metrics to utilise in our own custom filtering, which is once again provided by the DropletUtils tool.
Hands-on: Custom Filtering
DropletUtilsTool: toolshed.g2.bx.psu.edu/repos/iuc/dropletutils/dropletutils/1.10.0+galaxy1 with the following parameters:
“Format for the input matrix”: Bundled (barcodes.tsv, genes.tsv, matrix.mtx)
param-file“Count Data”: Matrix Gene Counts (output of RNA STARsolotool)
param-file“Genes List”: Genes (output of RNA STARsolotool)
param-file“Barcodes List”: Barcodes (output of RNA STARsolotool)
“Operation”: Filter for Barcodes
“Method”: EmptyDrops
“Lower-bound Threshold”: 200
“FDR Threshold”: 0.01
“Format for output matrices”: Tabular
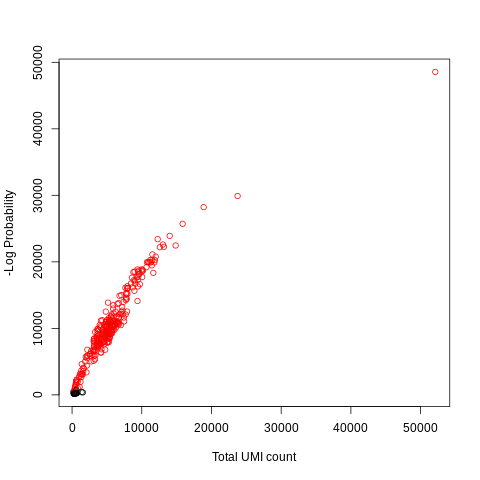
Figure 8: Detected Cells (red)
Here we recover 278 high quality cells instead of the 272 detected via the default method previously. On large datasets, this difference can help clean downstream clustering. For example, soft or less well-defined clusters are derived from too much noise in the data due to too many low quality cells being in the data during the clustering. Filtering these out during the pre-processing would produce much better separation, albeit at the cost of having less cells to cluster. This filter-cluster trade-off is discussed in more detail in the downstream analysis training materials.
Conclusion
In this workflow we have learned to quickly perform mapping and quantification of scRNA-seq FASTQ data in a single step via RNA STARsolo, and have reproduced a Cell Rangerworkflow using the DropletUtils suite, where we further explored the use of barcode rankings to determine better filtering thresholds to generate a high quality count matrix.
A full pipeline which produces both an AnnData and tabular file for inspection is provided here.
Note that, since version 2.7.7a of the tool, the entire Cell Ranger pipeline including the filtering can be performed natively within RNA STARsolo. As this is still a relatively new feature, we do not use it here in this tutorial, but eager users are encouraged to try it out.
This tutorial is part of the https://singlecell.usegalaxy.eu portal (Tekman et al. 2020).
Key points
Barcode FASTQ Reads are used to parse cDNA sequencing Reads.
A raw matrix is too large to process alone, and need to be filtered into a high quality one for downstream analysis
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
References
Tekman, M., B. Batut, A. Ostrovsky, C. Antoniewski, D. Clements et al., 2020 A single-cell RNA-sequencing training and analysis suite using the Galaxy framework. GigaScience 9: giaa102.
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.
Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{transcriptomics-scrna-preprocessing-tenx,
author = "Mehmet Tekman and Hans-Rudolf Hotz and Daniel Blankenberg and Wendi Bacon",
title = "Pre-processing of 10X Single-Cell RNA Datasets (Galaxy Training Materials)",
year = "2022",
month = "10",
day = "18"
url = "\url{https://training.galaxyproject.org/training-material/topics/transcriptomics/tutorials/scrna-preprocessing-tenx/tutorial.html}",
note = "[Online; accessed TODAY]"
}
@article{Batut_2018,
doi = {10.1016/j.cels.2018.05.012},
url = {https://doi.org/10.1016%2Fj.cels.2018.05.012},
year = 2018,
month = {jun},
publisher = {Elsevier {BV}},
volume = {6},
number = {6},
pages = {752--758.e1},
author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning},
title = {Community-Driven Data Analysis Training for Biology},
journal = {Cell Systems}
}
Congratulations on successfully completing this tutorial!
Do you want to extend your knowledge? Follow one of our recommended follow-up trainings:
 Mehmet Tekman
Mehmet Tekman
 Hans-Rudolf Hotz
Hans-Rudolf Hotz Daniel Blankenberg
Daniel Blankenberg Wendi Bacon
Wendi Bacon Questions:
Questions: