Evaluating and ranking a set of pathways based on multiple metrics

 Kenza Bazi-Kabbaj
Kenza Bazi-Kabbaj Thomas Duigou
Thomas Duigou Joan Hérisson
Joan Hérisson Guillaume Gricourt
Guillaume Gricourt Ioana Popescu
Ioana Popescu Jean-Loup Faulon
Jean-Loup FaulonOverviewQuestions:Objectives:
How to evaluate a set of heterologous pathways ?
Requirements:
Calculate the production flux of the lycopene target molecule using Flux Balance Analysis tool.
Compute thermodynamics values to optimize the yield of the reaction producing the lycopene using Thermo tool.
Compute the global score for the previous annotated pathways.
Rank the computed heterologous pathways depending on their score.
Time estimation: 20 minutesSupporting Materials:Last modification: Oct 18, 2022
 Questions:
Questions:
Introduction
Progress in synthetic biology is enabled by powerful bioinformatics tools such as those aimed to design metabolic pathways for the production of chemicals. These tools are available in SynBioCAD portal which is the first Galaxy set of tools for synthetic biology and metabolic engineering (Hérisson et al. 2022).
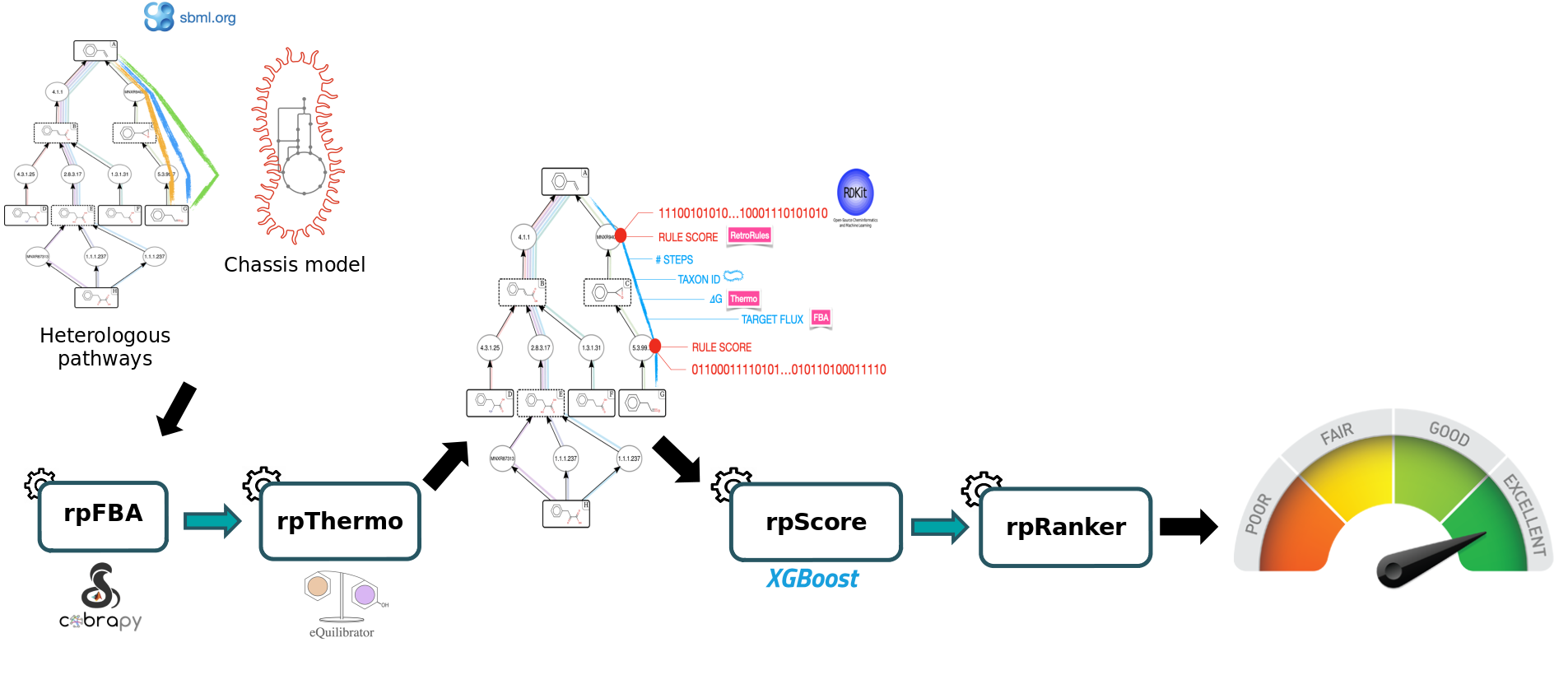
In this tutorial, we will use a set of tools from the Pathway Analysis workflow which will enable you to evaluate a set of heterelogous pathways previously produced by the RetroSynthesis workflow in a chassis organism (E. coli iML1515). These workflows are available in the Galaxy SynbioCAD platform. The goal is to inform the user of the theoretically best performing pathways by ranking them based on the four following criteria: target product flux, thermodynamic feasibility, pathway length and enzyme availability.
We recommend that you follow the Retrosynthesis tutorial before starting the current tutorial which will enable you to find pathways to synthesize heterologous compounds producing Lycopene in the E. coli chassis organism.
Four main steps will be run using the following workflow:
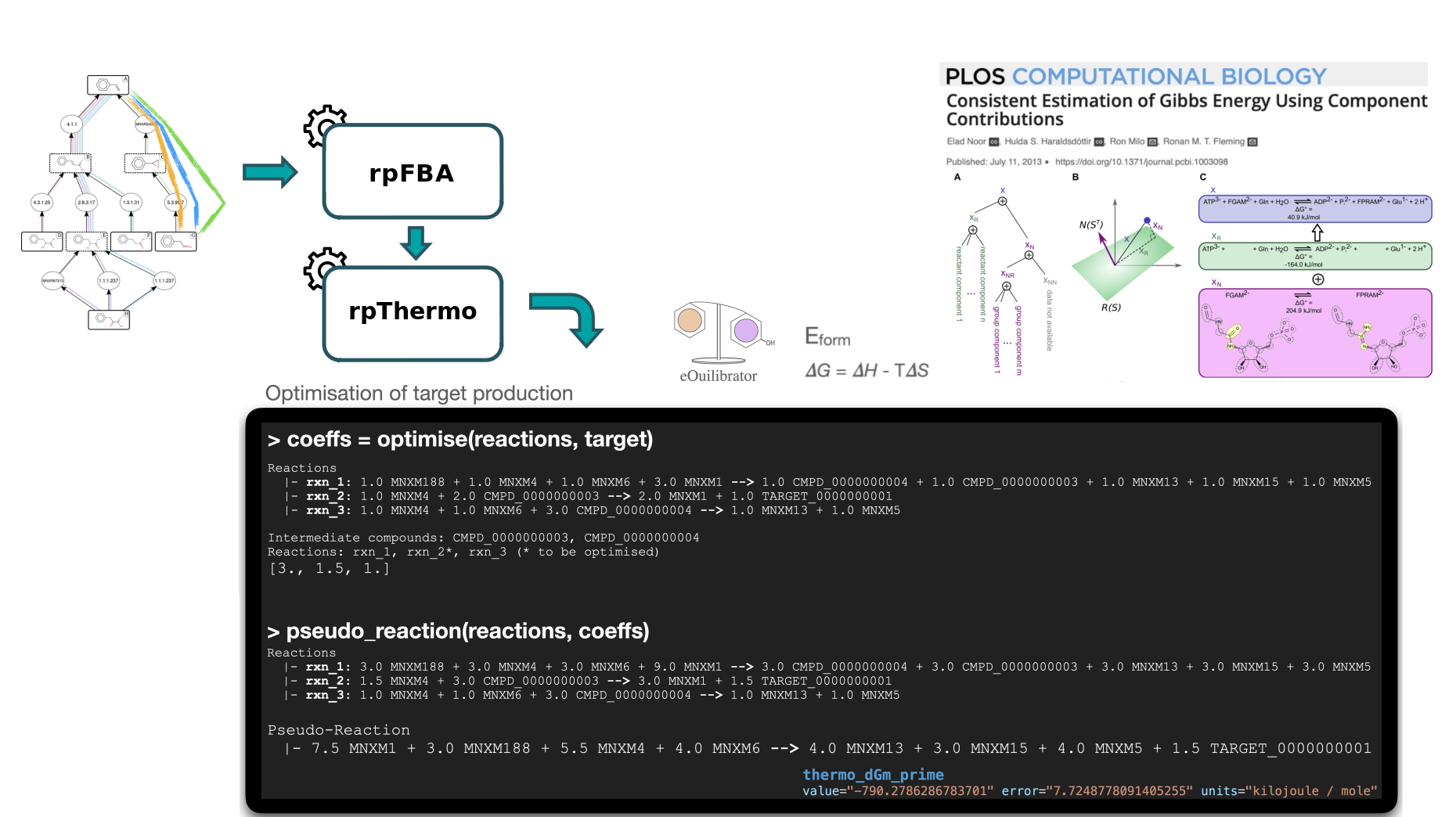
To rank the computed heterologous pathways, we need to calculate some metrics. This is why an in-house Flux Balance Analysis (FBA) was developed to calculate the production flux of a given target (e.g. lycopene). The method forces a fraction of its maximal flux through the biomass reaction while optimizing for the target molecule. This is achieved by the Flux Balance Analysis tool.
Secondly, we will use the Thermo tool to estimate thermodynamics values (based on Gibbs free energies) for each pathway to know whether a producing pathway is feasible in physiological conditions. The contribution of individual reactions to the final pathway thermodynamic is balanced solving a linear equation system.
After that, the Score Pathway tool is used to calculate a global score combining target flux, pathway thermodynamics, pathway length and enzyme availability.
Finally, the pathway are ranked based on the global score using the Rank Pathways tool.
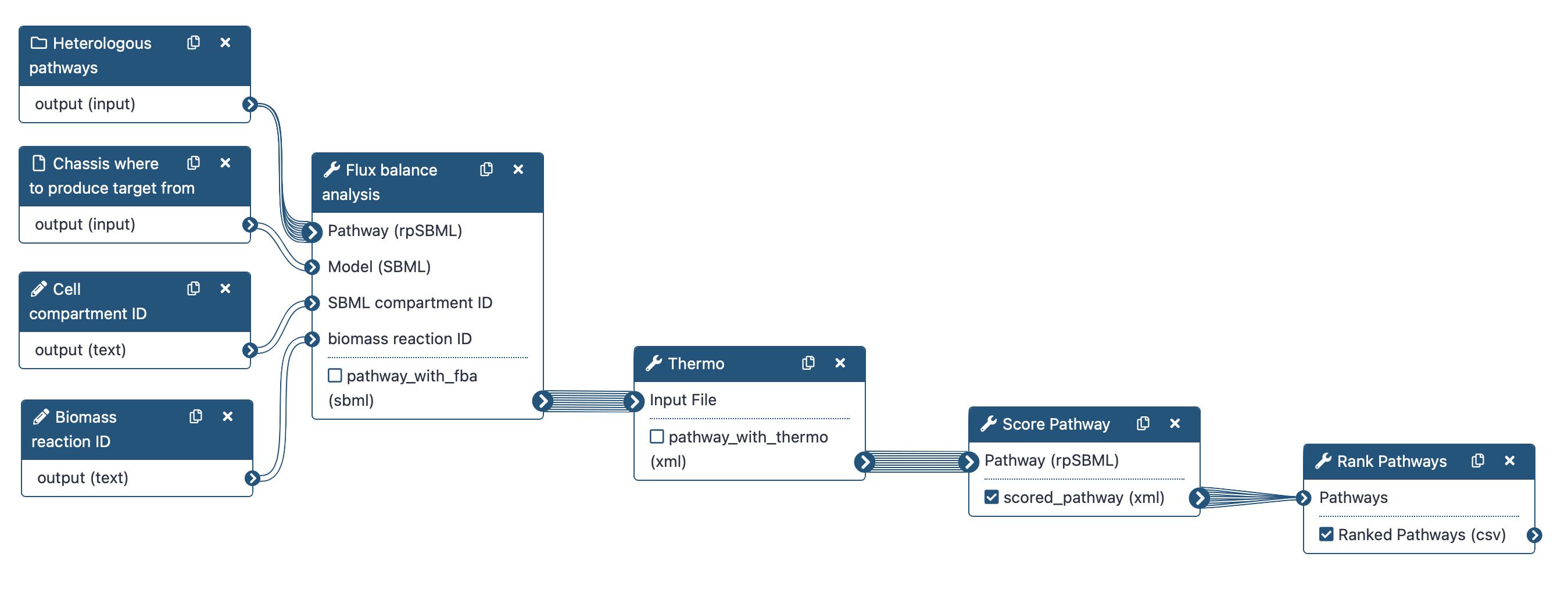
Note that we will run the steps of this workflow individually so as not to neglect the understanding of the intermediate steps. Then, we will run the workflow automatically so that it itself retrieves the outputs from the previous step and gives them as input to the next tool.
AgendaIn this tutorial, we will cover:
Data Preparation
First we need to upload and prepare the following inputs to analyze:
-
A set of pathways provided in the SBML format (Systems Biology Markup Language) to be ranked, modeling heterologous pathways such as those outputted by the RetroSynthesis workflow (available in Galaxy SynbioCAD platform).
-
The GEM (Genome-scale Metabolic Models) which is a formalized representation of the metabolism of the host organism (the model is E. coli iML1515), provided in the SBML format.
Get data
Hands-on: Data upload
- Create a new history for this tutorial named Pathway Analysis.
Import the files from Zenodo :
https://zenodo.org/api/files/5db78fa1-b8cb-4046-b57c-8a9d00806f42/rp_001_0001.xml https://zenodo.org/api/files/5db78fa1-b8cb-4046-b57c-8a9d00806f42/rp_001_0006.xml https://zenodo.org/api/files/5db78fa1-b8cb-4046-b57c-8a9d00806f42/rp_001_0011.xml https://zenodo.org/api/files/5db78fa1-b8cb-4046-b57c-8a9d00806f42/rp_002_0001.xml https://zenodo.org/api/files/5db78fa1-b8cb-4046-b57c-8a9d00806f42/rp_002_0006.xml https://zenodo.org/api/files/5db78fa1-b8cb-4046-b57c-8a9d00806f42/rp_002_0011.xml https://zenodo.org/api/files/5db78fa1-b8cb-4046-b57c-8a9d00806f42/rp_003_0001.xml https://zenodo.org/api/files/5db78fa1-b8cb-4046-b57c-8a9d00806f42/rp_003_0116.xml https://zenodo.org/api/files/5db78fa1-b8cb-4046-b57c-8a9d00806f42/rp_003_0231.xml https://zenodo.org/api/files/5db78fa1-b8cb-4046-b57c-8a9d00806f42/SBML_Model_iML1515.xml
- Copy the link location
Open the Galaxy Upload Manager (galaxy-upload on the top-right of the tool panel)
- Select Paste/Fetch Data
Paste the link into the text field
Press Start
- Close the window
Create a list or collection named
Heterologous pathwaysand composed of the 9 rpSBML pathways.
- Click on Operations on multiple datasets (check box icon) at the top of the history panel
- Check all the datasets in your history you would like to include
Click For all selected.. and choose Build dataset list
- Enter a name for your collection
- Click Create List to build your collection
- Click on the checkmark icon at the top of your history again
Compute the target product flux
Notice that the starting compounds (in other words, the precursors) of the predicted pathways (also referred as the heterologous pathways) are compounds that have been initially extracted from the genome-scale metabolic model (GEM) of the organism we are interested in (also referred as chassis). While this step is out of the scope of the present Pathway Analysis tutorial, this means that the precursors of predicted pathways are also present in the chassis model. Hence, predicted pathways and the chassis organism model can be merged to construct “augmented” whole-cell models, enabling flux analysis of these metabolic systems. This is what we’ll do here to predict the production flux of a compound of interest.
Within the frame of this tutorial, we’ll use the E. coli iML1515 GEM (downloaded from the BiGG database) to model the chassis metabolism of E. coli and the target compound is the lycopene. The provided E. coli model is in the SBML. The extraction of precursor compounds and the pathway prediction have already been performed during the RetroSynthesis workflow (available in Galaxy SynbioCAD platform).
The FBA (Flux Balance Analysis) method used to calculate the flux is a mathematical approach (as decribed in section Methods in Hérisson et al. 2022) which uses the COBRApy package (Ebrahim et al. 2013) and proposes 3 different analysis methods (standard FBA, parsimonious FBA, fraction of reaction). The first two methods are specific to the COBRApy package and the last one Fraction of Reaction is an in-house analysis method (as decribed in section Methods in Hérisson et al. 2022) to consider the cell needs for its own maintenance while producing the target compound.
Within the workflow, the purpose of the Flux Balance Analysis tool is to predict the production flux of the targeted compound, while considering the cellular needs. Under such simulation conditions, the analysis that returns a low production flux may be due to some precursor compounds having a limiting production flux, nor cofactor fluxes involved not being sufficiently balanced by the chassis native metabolism. Pathways with high flux would be caused by both the precursor compounds and the cofactors being in abundance. In either case, bottlenecks that limit the flux of the pathway may be investigated (this is outside of the scope of the workflow) and pathways that do not theoretically generate high yields can be filtered out.
We first perform an FBA (with COBRApy) optimizing the biomass reaction and record its maximal theoretical flux. The upper and lower bounds of the biomass reaction are then set to a same amount, equals to a fraction of its previously recorded optimum (default is 75% of its optimum). The method then performs a second FBA where biomass flux is enforced to this fraction of its optimum while optimizing the target production flux. Simulated fluxes are recorded directly into the SBML file and all changed flux bounds are reset to their original values before saving the output file.
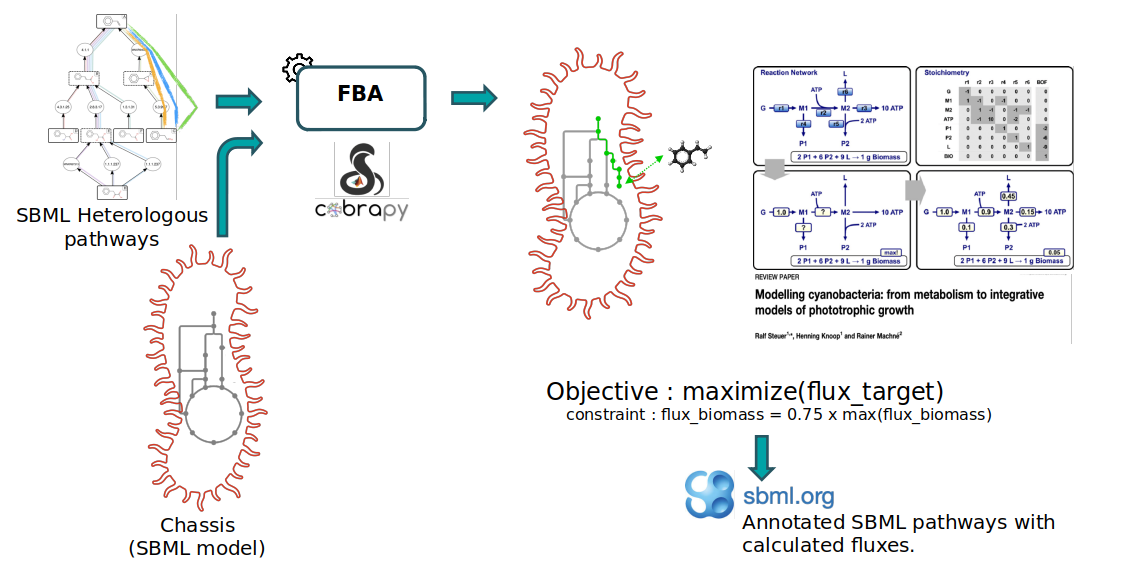
Blocking compounds that cannot provide any flux are temporarily removed from heterologous reactions for the FBA evaluation. Such cases can happen due to side substrates or products of predicted reactions that do not match any chassis compound, representing dead-end paths.
Hands-on: Calculating the flux of a target using Flux Balance Analysis (FBA)
- Run Flux balance analysis Tool: toolshed.g2.bx.psu.edu/repos/iuc/rpfba/rpfba/5.12.1 with the following parameters:
- param-collection “Pathway (rpSBML)”: Select
Heterologous pathways(Input dataset collection) from your current history.- param-file “Model (SBML)”: Select
SBML_Model_iML1515.xml(Input dataset) from your current history.- “SBML compartment ID”: Leave the default value
c.Comment: Choose a compartment corresponding to your modelYou can specify the compartment from which the chemical species were extracted. The default is
c, the BiGG code for the cytoplasm.
- “biomass reaction ID”: Specify the biomass reaction ID that will be used for the “fraction” simulation, type
R_BIOMASS_Ec_iML1515_core_75p37M.Comment: How to select the biomass reaction ID ?The biomass reaction ID objective is extracted from the current model E.Coli iML1515. You can search the term
biomassin your XML model and pick the ID where the termcoreappears.
- “Constraint based simulation type”:
Fraction of Reaction
Question
- What is the FBA score for
rp_003_0001pathway ?
- View the SBML rp_003_0001 file and look for
fba_fractionvalue in<groups:listOfGroups>section: value=0.23693089430893874.
Compute thermodynamics values
The goal of the thermodynamic analysis is to estimate the feasibility of the predicted pathways toward target production, in physiological conditions. The eQuilibrator libraries (Flamholz et al. 2011) are used to calculate the formation energy of compounds by either using public database IDs (when referenced within the tools internal database) or by decomposing the chemical structure and calculating its energy of formation using the component contribution method.
The reaction Gibbs energy is estimated by combining the energy of formation of the compounds involved in the reaction (with consideration for the stoichiometric coefficients).
Finally, the thermodynamic of a pathway is estimated by combining the Gibbs energy of reactions involved in it. The contribution of individual reactions to the final pathway thermodynamic is balanced using a linear equation system, according to the relative uses of intermediate compounds across the pathway (See Thermodynamics in Methods section for further details: Hérisson et al. 2022). A pathway Gibbs energy below zero indicates that the thermodynamic is favorable toward the production of the target.
Secondly, we will use the Thermo tool to estimate thermodynamics values (based on Gibbs free energies) for each pathway to know whether a producing pathway is feasible in physiological conditions
Hands-on: Compute thermodynamics values for each pathway using rpThermo tool
- Thermo Tool: toolshed.g2.bx.psu.edu/repos/tduigou/rpthermo/rpthermo/5.12.1 with the following parameters:
- param-file “Input File”:
pathway_with_fba(output of Flux balance analysis tool)CommentThe tool takes as input pathways in SBML format and returns annotated pathways (with thermodynamics information for each reaction) in SBML format too.
Question
- What is the thermodynamic score attributed to the reaction with the following EC (Enzyme Commission) number 2.5.1.29 for
rp_001_0001pathway ?
- View the SBML rp_001_0001 file and search the reaction ID
2.5.1.29contained in<listOfReactions>. The corresponding value is indicated inthermo_dGm_primefield :-242.348.
Compute the global score of pathways
The Pathway Score tool provides a global score for a given pathway previously annotated by the Flux Balance Analysis and Thermo tools. This score is computed by a machine learning (ML) model (cf. Machine Learning Global Scoring in Hérisson et al. 2022). The model takes as input features describing the pathway (thermodynamic feasibility, target flux with fixed biomass, length) and the reactions within the pathway (reaction SMARTS, Gibbs free energy, enzyme availability score) and prints out the probability for the pathway to be a valid pathway. The ML model has been trained on literature data (cf. section Benchmarking with literature data in Hérisson et al. 2022) and by a validation trial (cf. section Benchmarking by expert validation trial in Hérisson et al. 2022).
Hands-on: Compute the global score using the _Pathway Score_ tool
- Score Pathway Tool: toolshed.g2.bx.psu.edu/repos/tduigou/rpscore/rpscore/5.12.1 with the following parameters:
- param-file “Pathway (rpSBML)”:
pathway_with_thermo(output of Thermo tool)CommentThis tool will output a new annotated SBML file representing the pathway, containing the
global_scoreannotation.
Question
- What is the computed global score for the
rp_001_0001pathway ?
- View the SBML file
rp_001_0001and search forglobal_score: value=0.975147980451584.
Rank annotated pathways
Finally, Rank Pathways ranks the previous set of heterologous pathways, based on their global score, to reveal what are the most likely pathways to produce the target molecule (here it is lycopene) in a given organism of interest (E. coli in this tutorial).
Hands-on: Rank annotated pathways using rpRanker tool
- Rank Pathways Tool: toolshed.g2.bx.psu.edu/repos/tduigou/rpranker/rpranker/5.12.1 with the following parameters:
- param-file “Pathways”:
scored_pathway(output of Score Pathway tool)CommentThis tool will output a CSV file which contains the pathway IDs and their corresponding global score.
Question
- What are the 3 top-ranked pathways ?
002_0011,001_0011,002_0006.
Run the Pathway Analysis Workflow
In this section, you can run the Pathway Analysis Workflow more easily and fastly following these instructions:
Hands-on: Execute the entire workflow in one go.
Import your Pathway Analysis Workflow by uploading the workflow file.
- Click on Workflow on the top menu bar of Galaxy. You will see a list of all your workflows.
- Click on the upload icon galaxy-upload at the top-right of the screen
- Provide your workflow
- Option 1: Paste the URL of the workflow into the box labelled “Archived Workflow URL”
- Option 2: Upload the workflow file in the box labelled “Archived Workflow File”
- Click the Import workflow button
- Click on Workflow on the top menu bar of Galaxy. You will see Pathway Analysis Workflow
- Click on the workflow-run (Run workflow) button next to your workflow
- Provide the workflow with the following parameters:
- param-file “Heterologous pathways”: Select
Heterologous pathways(Input dataset collection) from your current history.- param-file “Chassis where to produce target from”: Select
SBML_Model_iML1515.xml(Input dataset) from your current history.- “Cell compartment ID”: Enter value
c.- “biomass reaction ID”: Specify the biomass reaction ID that will be restricted in the “fraction” simulation type
R_BIOMASS_Ec_iML1515_core_75p37M.CommentAll the outputs will be automatically generated and identical to the previous ones.
Conclusion
To select the best pathways for producing the lycopene in E. coli, some metrics have to be estimated, namely production flux of the target and pathway thermodynamics. A global score is then computed by combining these criteria with others (pathway length, enzyme availability score, reaction SMARTS) using a machine learning model. These steps achieved using the tools of the presented Pathway Analysis workflow.
Frequently Asked Questions
Have questions about this tutorial? Check out the FAQ page for the Synthetic Biology topic to see if your question is listed there. If not, please ask your question on the GTN Gitter Channel or the Galaxy Help ForumReferences
- Flamholz, A., E. Noor, A. Bar-Even, and R. Milo, 2011 eQuilibrator–the biochemical thermodynamics calculator. Nucleic Acids Research 40: D770–D775. 10.1093/nar/gkr874
- Ebrahim, A., J. A. Lerman, B. O. Palsson, and D. R. Hyduke, 2013 COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Systems Biology 7: 10.1186/1752-0509-7-74
- Hérisson, J., T. Duigou, M. du Lac, K. Bazi-Kabbaj, M. S. Azad et al., 2022 Galaxy-SynBioCAD: Automated Pipeline for Synthetic Biology Design and Engineering. 10.1101/2022.02.23.481618
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Kenza Bazi-Kabbaj, Thomas Duigou, Joan Hérisson, Guillaume Gricourt, Ioana Popescu, Jean-Loup Faulon, 2022 Evaluating and ranking a set of pathways based on multiple metrics (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/synthetic-biology/tutorials/pathway_analysis/tutorial.html Online; accessed TODAY
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
Congratulations on successfully completing this tutorial!@misc{synthetic-biology-pathway_analysis, author = "Kenza Bazi-Kabbaj and Thomas Duigou and Joan Hérisson and Guillaume Gricourt and Ioana Popescu and Jean-Loup Faulon", title = "Evaluating and ranking a set of pathways based on multiple metrics (Galaxy Training Materials)", year = "2022", month = "10", day = "18" url = "\url{https://training.galaxyproject.org/training-material/topics/synthetic-biology/tutorials/pathway_analysis/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Batut_2018, doi = {10.1016/j.cels.2018.05.012}, url = {https://doi.org/10.1016%2Fj.cels.2018.05.012}, year = 2018, month = {jun}, publisher = {Elsevier {BV}}, volume = {6}, number = {6}, pages = {752--758.e1}, author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning}, title = {Community-Driven Data Analysis Training for Biology}, journal = {Cell Systems} }