Multisample Analysis
 Anton Nekrutenko
Anton Nekrutenko
 Anne Pajon
Anne Pajon
OverviewQuestions:Objectives:
Time estimation: 1 hourLevel: Advanced AdvancedLast modification: Oct 18, 2022
 Questions:
Questions:
AgendaIn this tutorial, we will deal with:
Processing many samples at once
Here we will show Galaxy features designed to help with the analysis of large numbers of samples. When you have just a few samples - clicking through them is easy. But once you’ve got hundreds - it becomes very annoying. In Galaxy we have introduced Dataset collections that allow you to combine numerous datasets in a single entity that can be easily manipulated.
In this tutorial we assume the following:
- you already have basic understanding of how Galaxy works (if you don’t see Galaxy 101 tutorial;
- you have an account in Galaxy (see this if you don’t);
- you have your browser configured as described here.
warning At this time this tutorial is using Galaxy’s test server at https://test.galaxyproject.org. Once the main site is updated this tutorial will be edited.
Getting data
Here is a history containing a few datasets we will be practicing with (as always with Galaxy tutorial you can upload your own data and play with it instead of the provided datasets):
M117-bl_1- family 117, mother, 1-st (F) read from bloodM117-bl_2- family 117, mother, 2-nd (R) read from bloodM117-ch_1- family 117, mother, 1-st (F) read from cheekM117-ch_1- family 117, mother, 2-nd (R) read from cheekM117C1-bl_1- family 117, child, 1-st (F) read from bloodM117C1-bl_2- family 117, child, 2-nd (R) read from bloodM117C1-ch_1- family 117, child, 1-st (F) read from cheekM117C1-ch_2- family 117, child, 2-nd (R) read from cheek
These datasets represent genomic DNA (enriched for mitochondria via a long range PCR) isolated from blood and cheek (buccal swab) of mother (M117) and her child (M117C1) that was sequenced on an Illumina miSeq machine as paired-read library (250-bp reads; see our 2014 manuscript for Methods).
Creating a list of paired datasets
If you imported history as described above, your screen will look something like this:
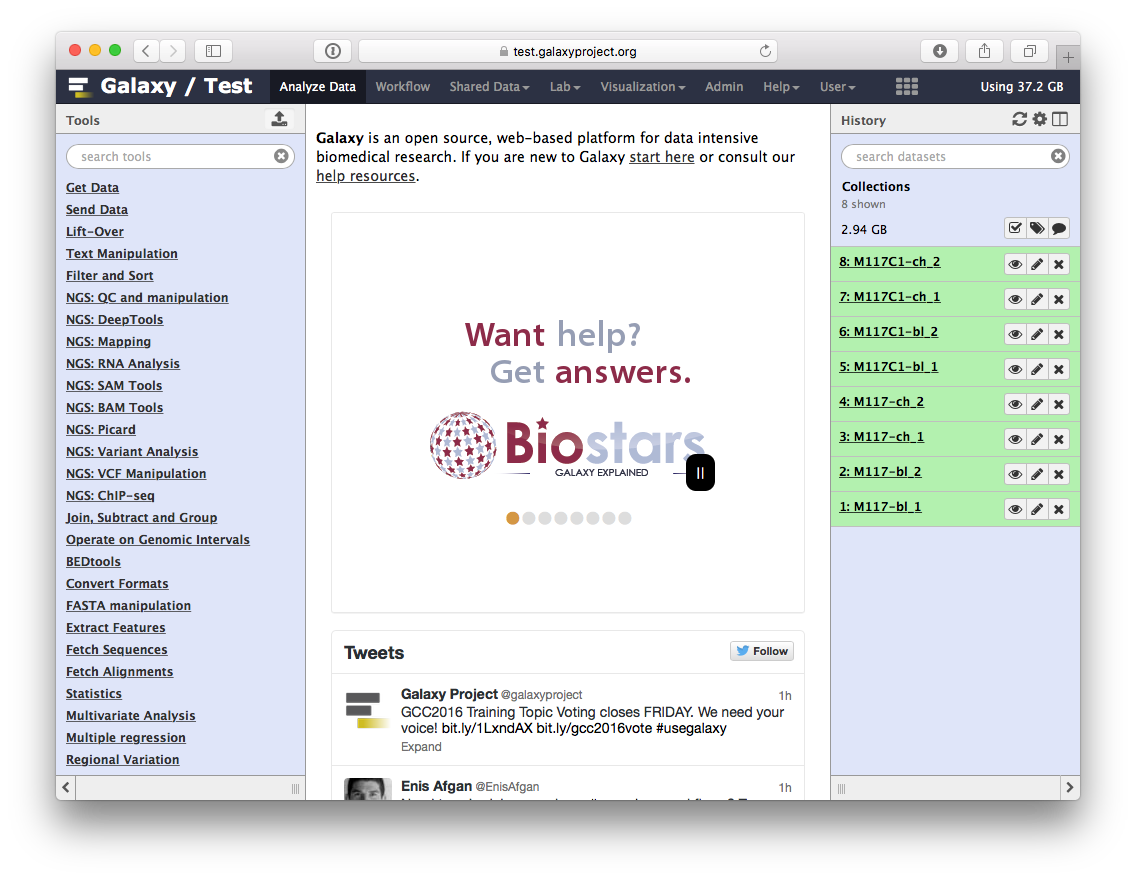
Now click the checkbox in  and you will see your history changing like this:
and you will see your history changing like this:
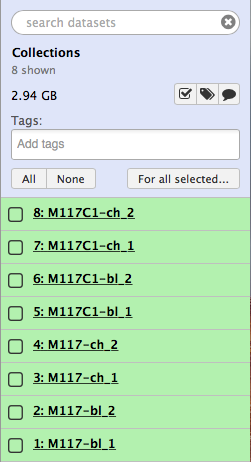
Let’s click All, which will select all datasets in the history, then click  and finally select Build List of Dataset Pairs from the following menu:
and finally select Build List of Dataset Pairs from the following menu:

The following wizard will appear:
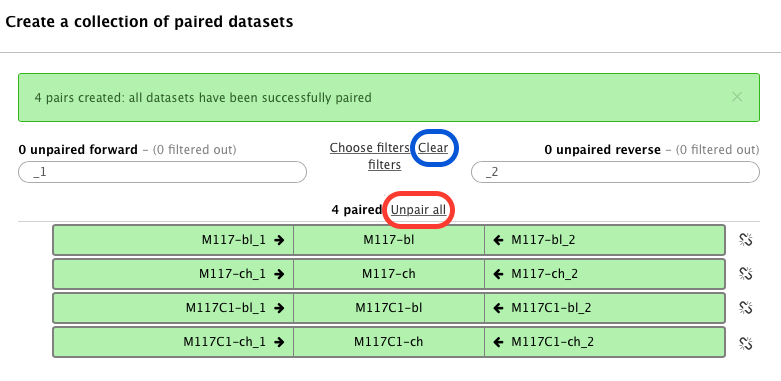
In this case Galaxy automatically assigned pairs using the _1 and _2 endings of dataset names. Let’s however pretend that this did not happen. Click on Unpair all (highlighted in red in the figure above) link and then on Clear link (highlighted in blue in the figure above). The interface will change into its virgin state:

Hopefully you remember that we have paired-end data in this scenario. Datasets containing the first (forward) and the second (reverse) read are differentiated by having _1 and _2 in the filename. We can use this feature in dataset collection wizard to pair our datasets. Type _1 in the left Filter this list text box and _2 in the right:

You will see that the dataset collection wizard will automatically filter lists on each side of the interface:
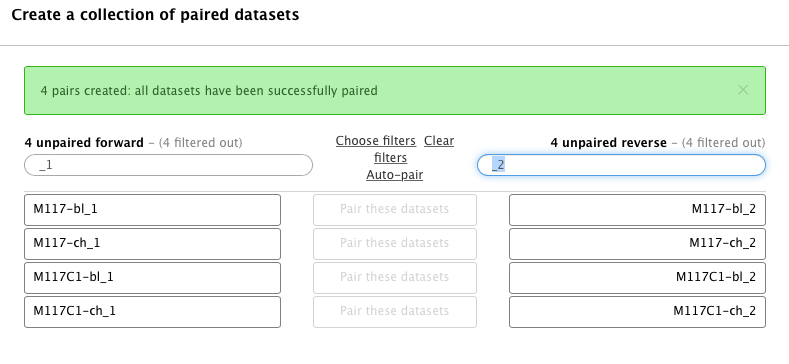
Now you can either click Auto pair if pairs look good to you (proper combinations of datasets are listed in each line) or pair each forward/reverse group individually by pressing Pair these datasets button separating each pair:
Now it is time to name the collection:
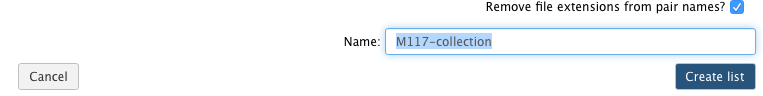
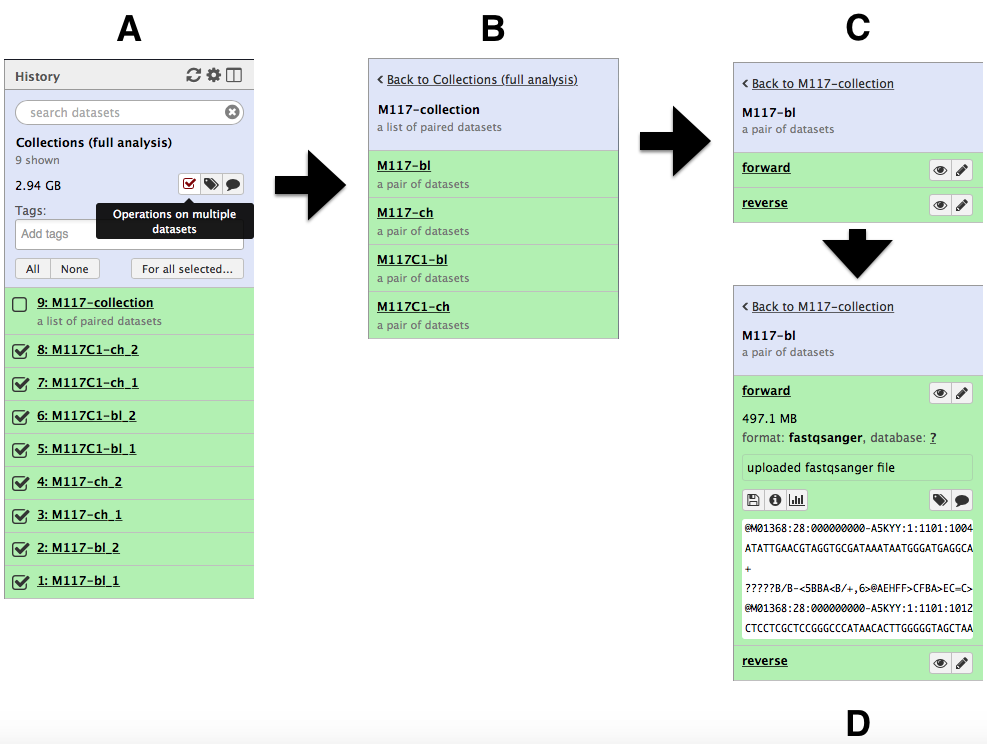
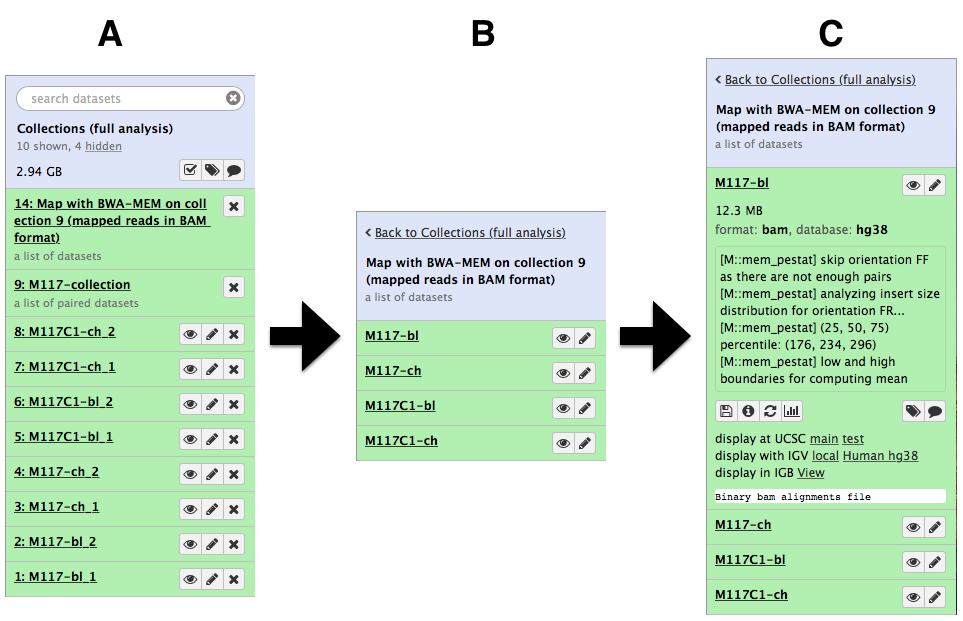
and create the collection by clicking Create list. A new item will appear in the history as you can see on the panel A below. Clicking on collection will expand it to show four pairs it contains (panel B). Clicking individual pairs will expand them further to reveal forward and reverse datasets (panel C). Expanding these further will enable one to see individual datasets (panel D).
Using collections
By now we see that a collection can be used to bundle a large number of items into a single history item. This means that many Galaxy tools will be able to process all datasets in a collection transparently to you. Let’s try to map these datasets to human genome using bwa-mem mapper:
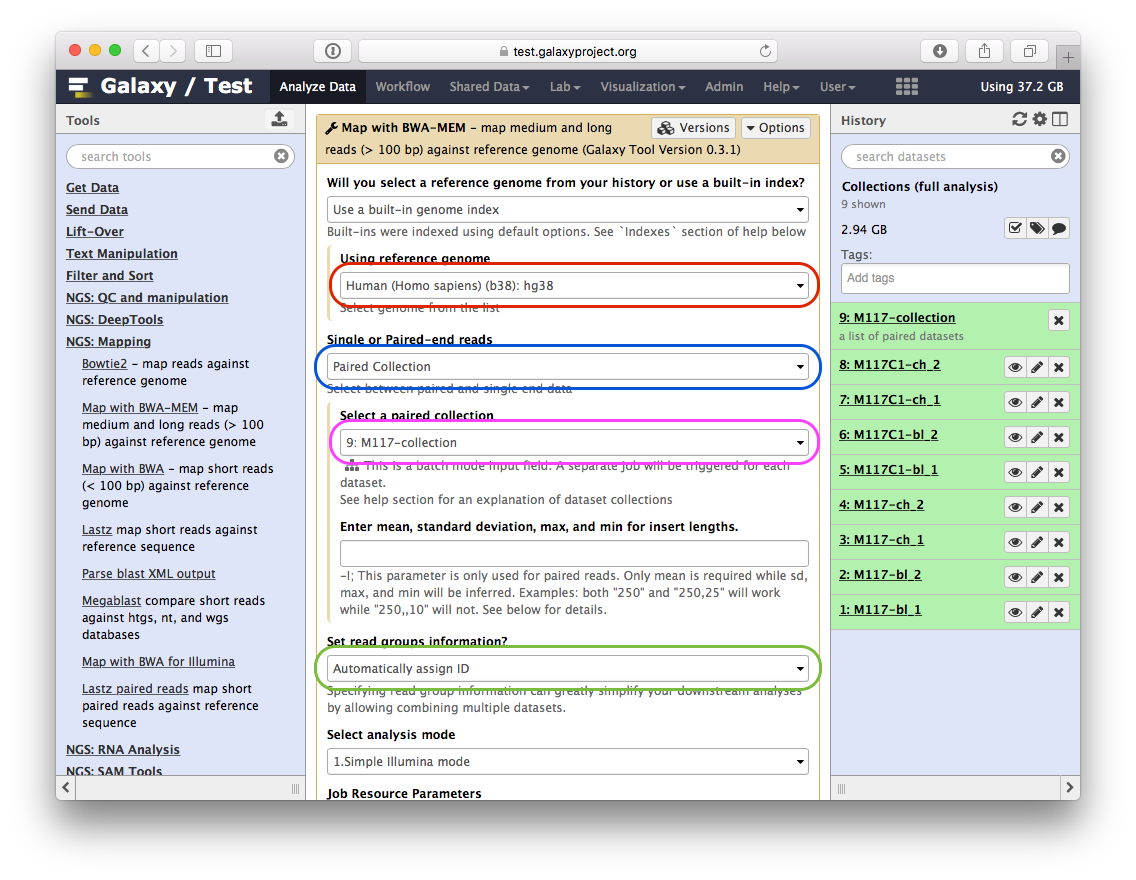
Here is what you need to do:
- set Using reference genome to
hg38(red outline); - set Single or Paired-end reads to
Paired collection(blue outline); - select
M177-collectionfrom Select a paired collection dropdown (magenta outline); - In Set read groups information select
Automatically assign ID(green outline); - scroll down and click Execute.
You will see jobs being submitted and new datasets appearing in the history. IN particular below you can see that Galaxy has started four jobs (two yellow and two gray). This is because we have eight paired datasets with each pair being processed separately by bwa-mem. As a result we have four bwa-mem runs:
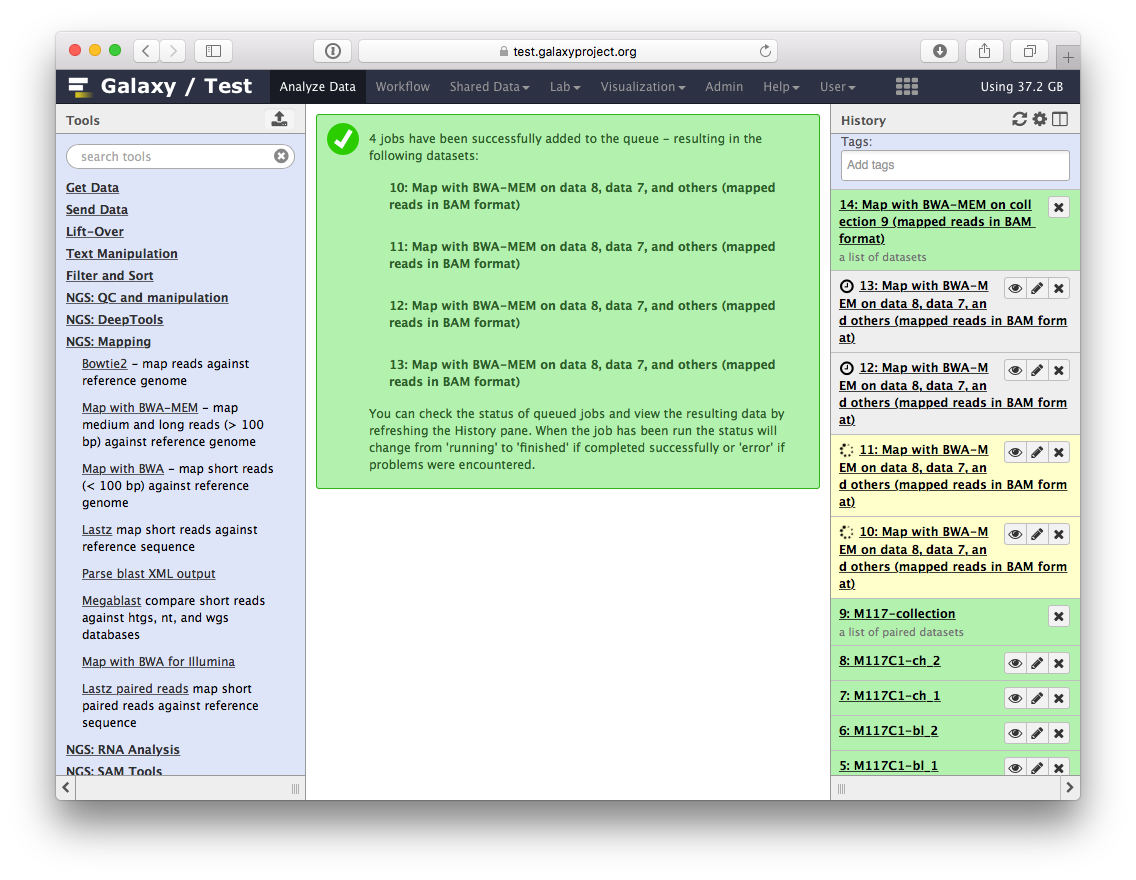
Once these jobs are finished they will disappear from the history and all results will be represented as a new collection:
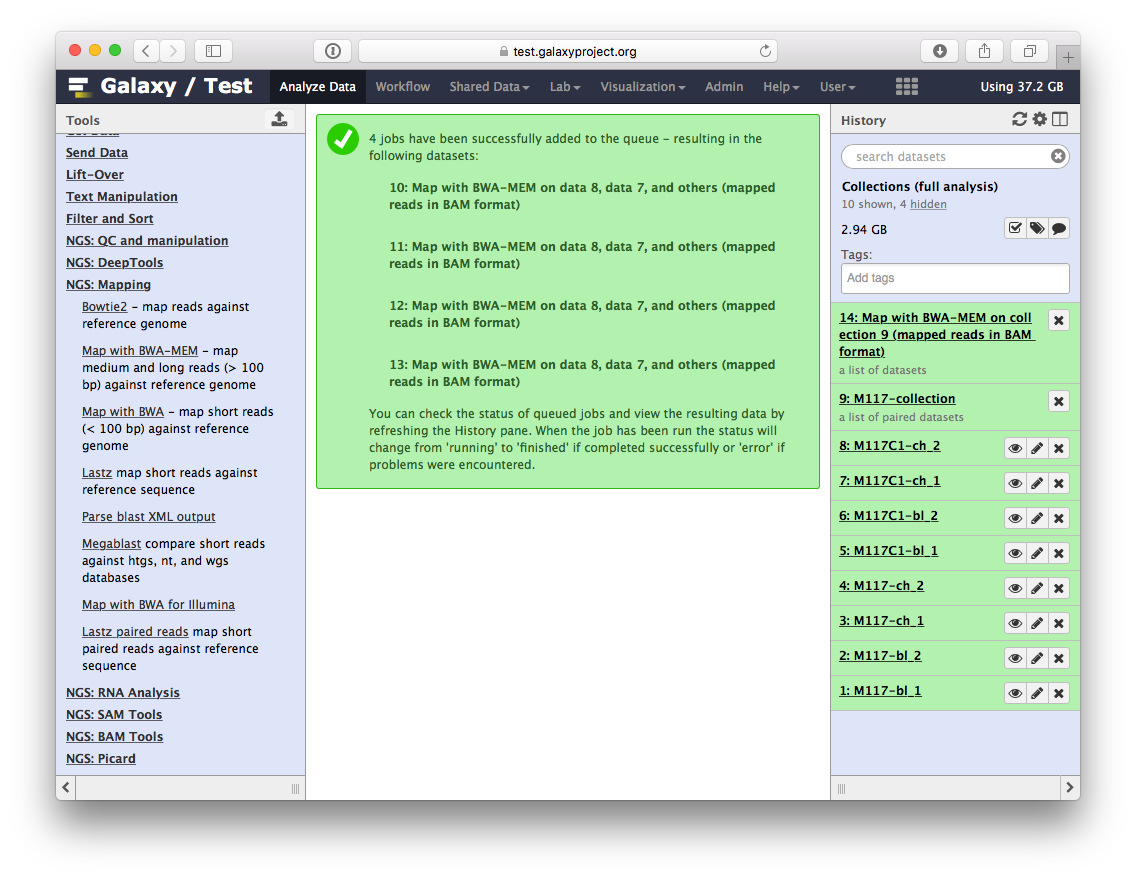
Let’s look at this collection by clicking on it (panel A in the figure below). You can see that now this collection is no longer paired (compared to the collection we created in the beginning of this tutorial). This is because bwa-mem takes forward and reverse data as input, but produces only a single BAM dataset as the output. So what we have in the result is a list of four dataset (BAM files; panels B and C).
Processing collection as a single entity
Now that bwa-mem has finished and generated a collection of BAM datasets we can continue to analyze the entire collection as a single Galaxy ‘item’.
Ensuring consistency of BAM dataset
Let’s perform cleanup of our BAM files with cleanSam utility from the Picard package:
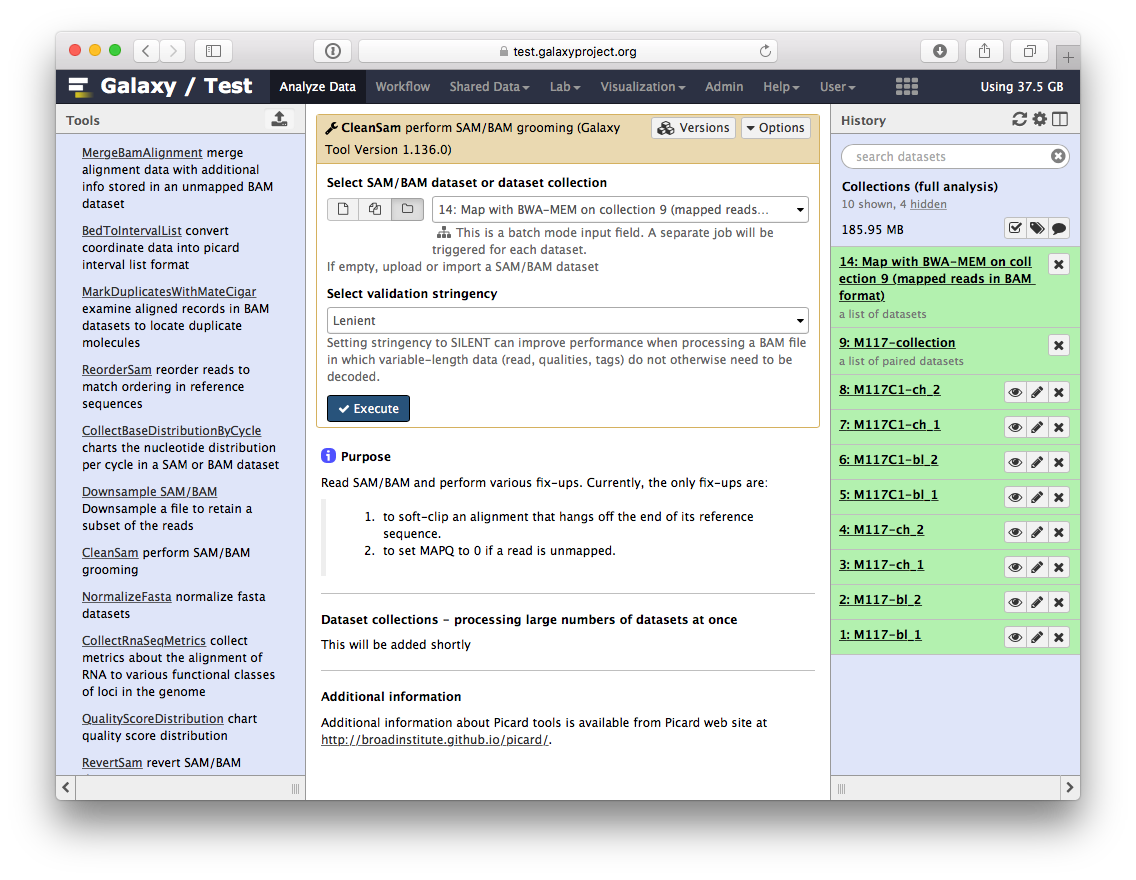
If you look at the picture above carefully, you will see that the Select SAM/BAM dataset or dataset collection parameter is empty (it says No sam or bam datasets available.). This is because we do not have single SAM or BAM datasets in the history. Instead we have a collection. So all you need to do is to click on the folder ( ) button and you will our BAM collection selected:
) button and you will our BAM collection selected:
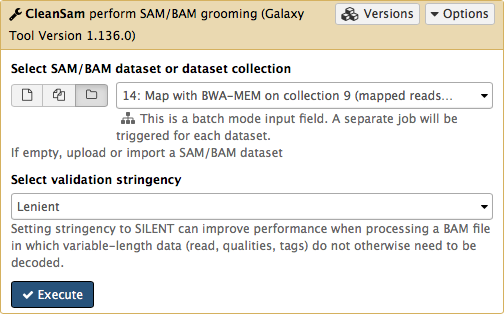
Click Execute. As an output this tool will produce a collection contained cleaned data.
Retaining ‘proper pairs’
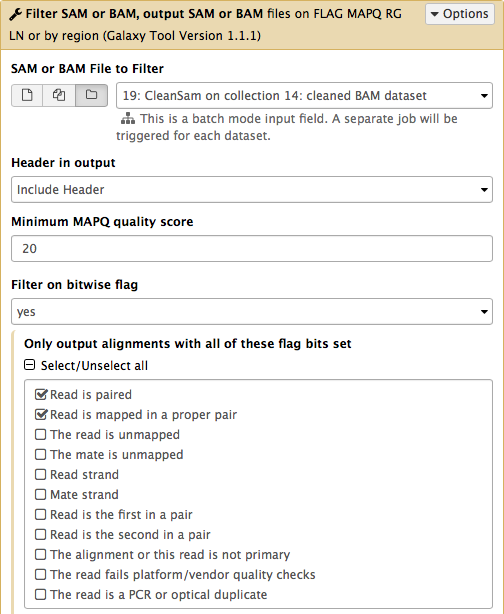
Now let’s clean the dataset further by only preserving truly paired reads (reads satisfying two requirements: (1) read is paired, and (2) it is mapped as a proper pair). For this we will use Filter SAM or BAM tools from SAMTools collection:
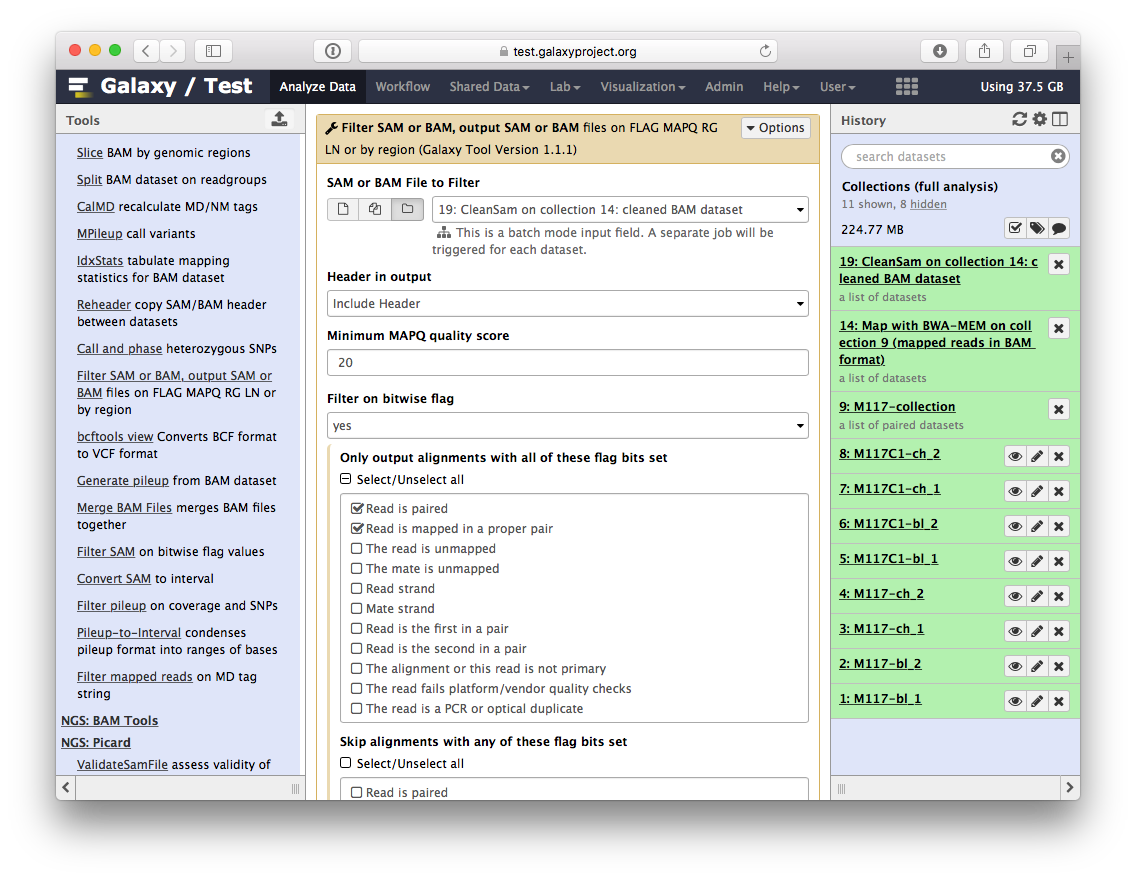
parameters should be set as shown below. By setting mapping quality to 20 we avoid reads mapping to multiple locations and by using Filter on bitwise flag option we ensure that the resulting dataset will contain only properly paired reads. This operation will produce yet another collection containing now filtered datasets.
Merging collection into a single dataset
The beauty of BAM datasets is that they can be combined in a single entity using so called Read group (learn more about Read Groups on old wiki, which will be migrated here shortly). This allows to bundle reads from multiple experiments into a single dataset where read identity is maintained by labelling every sequence with read group tags. So let’s finally reduce this collection to a single BAM dataset. For this we will use MergeSamFiles tool for the Picard suite:
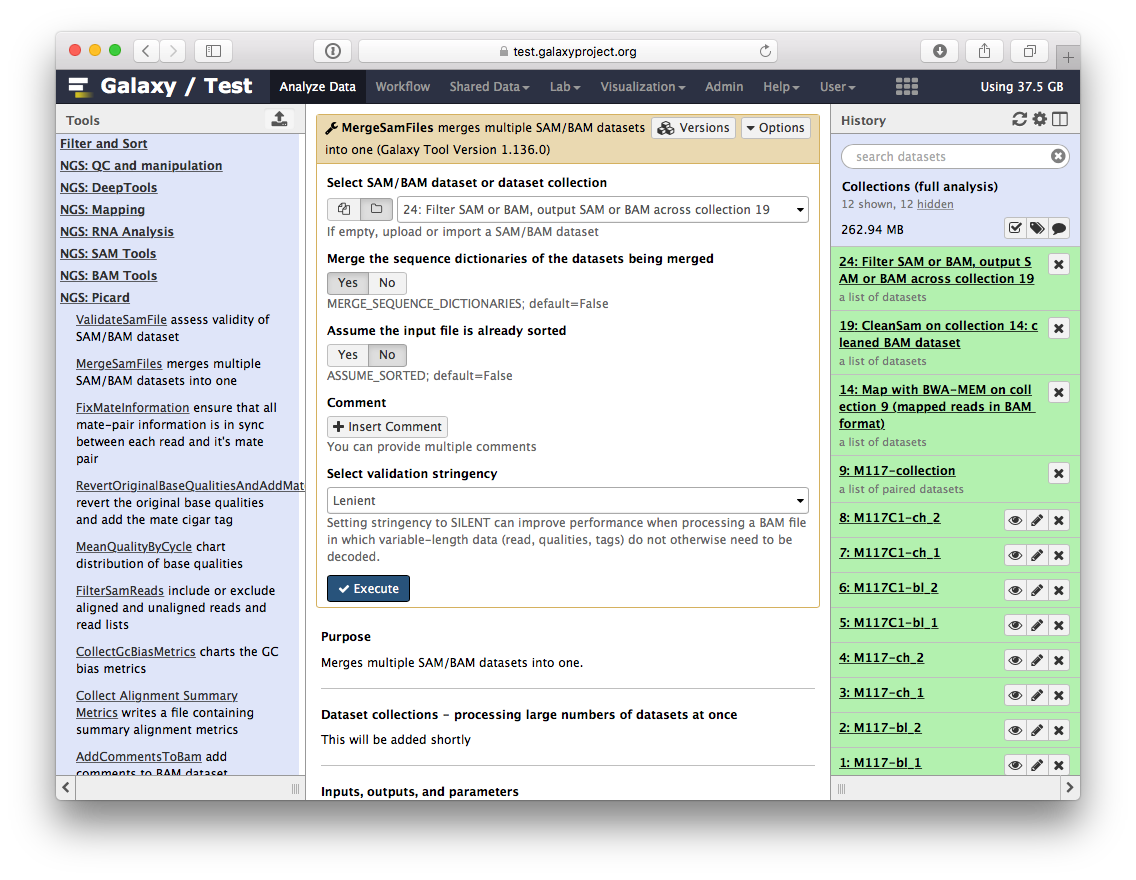
Here we select the collection generated by the filtering tool described above in 3.1:
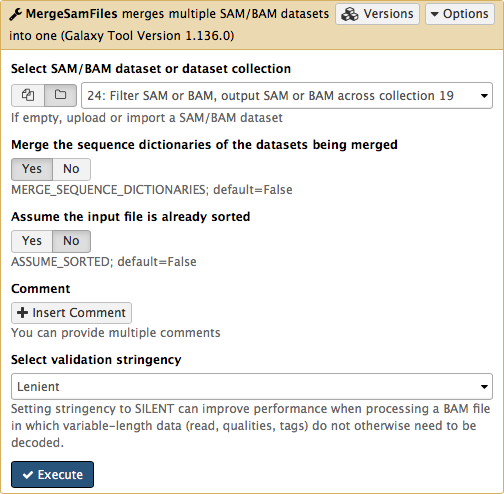
This operation will not generate a collection. Instead, it will generate a single BAM dataset containing mapped reads from our four samples (M117-bl, M117-ch, M117C1-bl, and M117C1-ch).
Let’s look at what we’ve got!
So we have one BAM dataset combining everything we’ve done so far. Let’s look at the contents of this dataset using a genome browser. First, we will need to downsample the dataset to avoiding overwhelming the browser. For this we will use Downsample SAM/BAM tool:
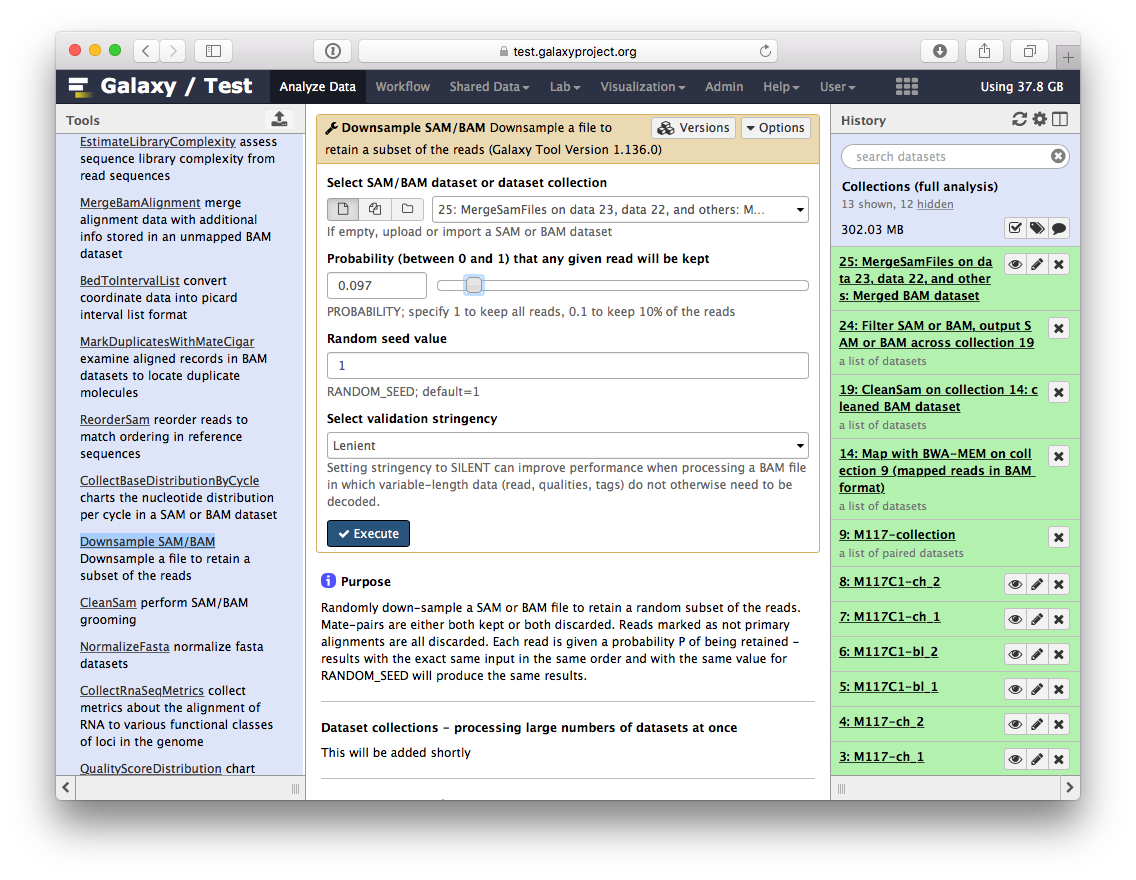
Set Probability (between 0 and 1) that any given read will be kept to roughly 5% (or 0.05) using the slider control:
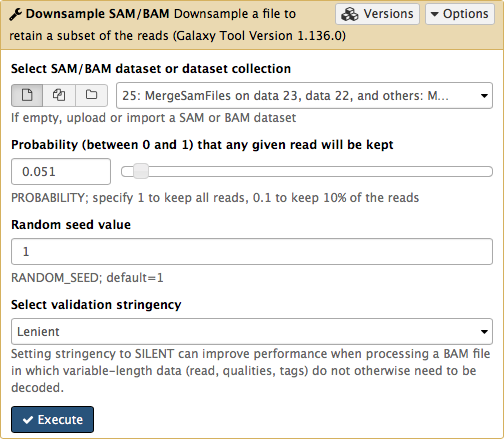
This will generate another BAM dataset containing only 5% of the original reads and much smaller as a result. Click on this dataset and you will see links to various genome browsers:
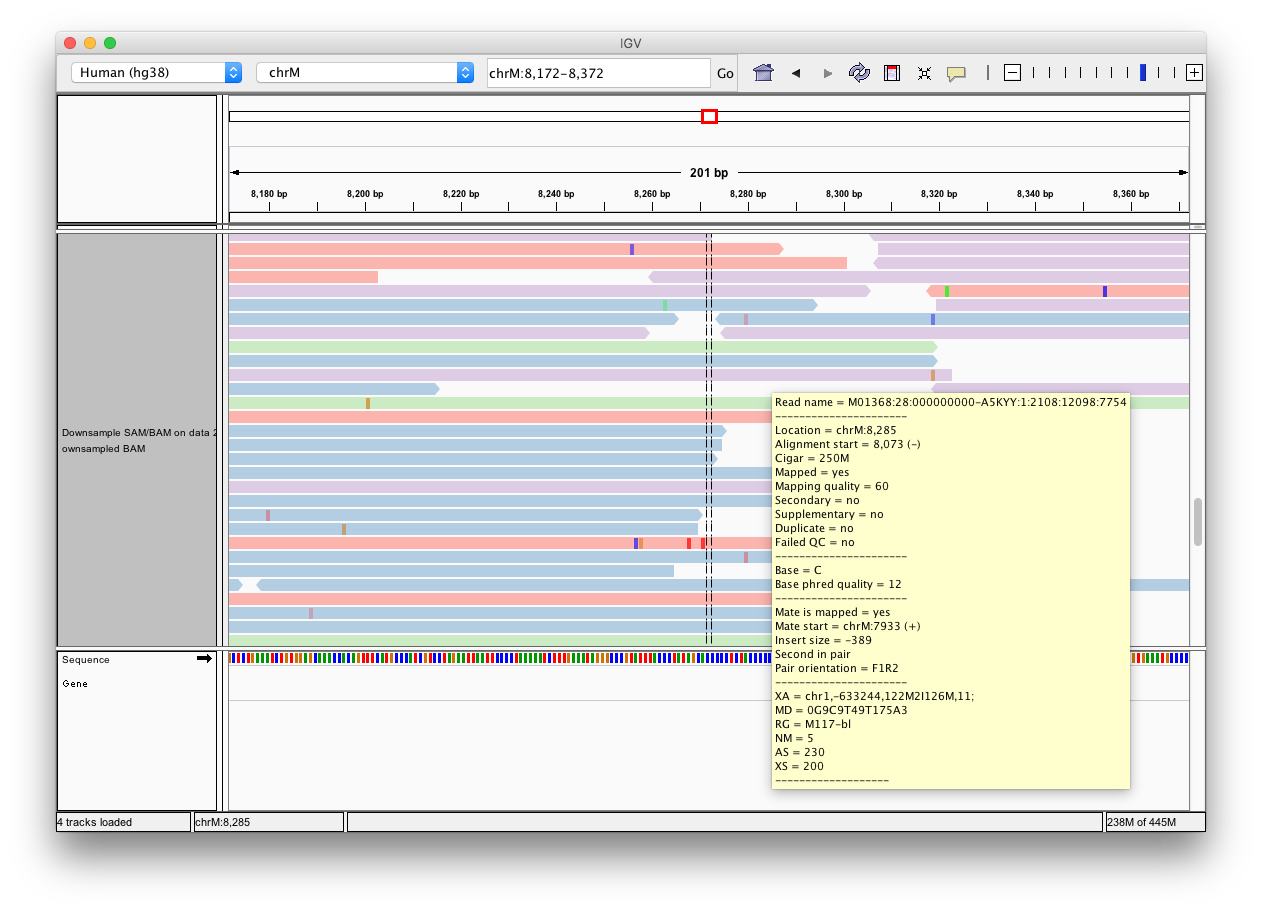
Click the Human hg38 link in the display with IGV line as highlighted above (learn more about displaying Galaxy data in IGV with this movie). Below is an example generated with IGV on these data. In this screenshot reads are colored by read group (four distinct colors). A yellow inset displays additional information about a single read. One can see that this read corresponds to read group M117-bl.
We did not fake this:
The two histories and the workflow described in this page are accessible directly from this page below:
- History Collections
- History Collections (full analysis)
From there you can import histories to make them your own.
If things don’t work…
…you need to complain. Use Galaxy’s Help Channel to do this.
Frequently Asked Questions
Have questions about this tutorial? Check out the FAQ page for the Using Galaxy and Managing your Data topic to see if your question is listed there. If not, please ask your question on the GTN Gitter Channel or the Galaxy Help ForumFeedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Anton Nekrutenko, Anne Pajon, 2022 Multisample Analysis (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/processing-many-samples-at-once/tutorial.html Online; accessed TODAY
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
Congratulations on successfully completing this tutorial!@misc{galaxy-interface-processing-many-samples-at-once, author = "Anton Nekrutenko and Anne Pajon", title = "Multisample Analysis (Galaxy Training Materials)", year = "2022", month = "10", day = "18" url = "\url{https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/processing-many-samples-at-once/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Batut_2018, doi = {10.1016/j.cels.2018.05.012}, url = {https://doi.org/10.1016%2Fj.cels.2018.05.012}, year = 2018, month = {jun}, publisher = {Elsevier {BV}}, volume = {6}, number = {6}, pages = {752--758.e1}, author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning}, title = {Community-Driven Data Analysis Training for Biology}, journal = {Cell Systems} }