The Vertebrate Genome Project (VGP), a project of the G10K Consortium, aims to generate high-quality, near error-free, gap-free, chromosome-level, haplotype-phased, annotated reference genome assemblies for every vertebrate species (Rhie et al. 2021). The VGP has developed a fully automated de-novo genome assembly pipeline, which uses a combination of three different technologies: Pacbio HiFi, Bionano optical maps and Hi-C chromatin interaction data.
As a result of a collaboration with the VGP team, a training including a step-by-step detailed description of parameter choices for each step of assembly was developed for the Galaxy Training Network (Lariviere et al. 2022). This tutorial instead provides a quick walkthrough on how the workflows can be used to rapidly assemble a genome using the VGP pipeline with the GalaxyWorkflow System (GWS).
GWS facilitates analysis repeatability, while minimizing the number of manual steps required to execute an analysis workflow, and automating the process of inputting parameters and software tool version tracking. The objective of this training is to explain how to run the VGP workflow, focusing on what are the required inputs and which outputs are generated and delegating how the steps are executed to the GWS.
This tutorial assumes you are comfortable getting data into Galaxy, running jobs, managing history, etc. If you are unfamiliar with Galaxy, we recommed you to visit the Galaxy Training Network. Consider starting with the following trainings:
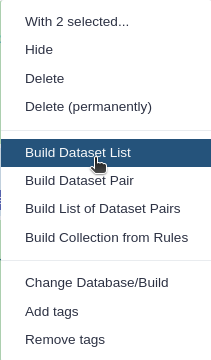
The VGP assembly pipeline has a modular organization, consisting in five main subworkflows (fig. 1), each one integrated by a series of data manipulation steps. Firstly, it allows the evaluation of intermediate steps, which facilitates the modification of parameters if necessary, without the need to start from the initial stage. Secondly, it allows to adapt the workflow to the available data.
Figure 1: VGP assembly pipeline. The VGP workflow is implemented in a modular fashion: it consists of five independent subworkflows. In addition, it includes some additional workflows (not shown in the figure), required for exporting the results to GenomeArk.
The VGP pipeline integrates two workflows to generate scaffolds from the contig level assemblies generated from the HiFi reads. When Hi-C data and Bionano data are available, the default pipeline is running the Bionano workflow first, followed by the Hi-C workflow. However, it is possible that Bionano data may not be available, in which case the HiC workflow can be used directly on the initial purged assembly.
Comment: Input option order
This tutorial assumes the input datasets are high-quality. QC on raw read data should be performed before it is used. QC on raw read data is outside the scope of this tutorial.
Get data
For this tutorial, the first step is to get the datasets from Zenodo. The VGP assembly pipeline uses data generated by a variety of technologies, including PacBio HiFi reads, Bionano optical maps, and Hi-C chromatin interaction maps.
Copy the tabular data, paste it into the textbox and press Build
dataset_01 https://zenodo.org/record/6098306/files/HiFi_synthetic_50x_01.fasta?download=1 fasta HiFi HiFi_collection
dataset_02 https://zenodo.org/record/6098306/files/HiFi_synthetic_50x_02.fasta?download=1 fasta HiFi HiFi_collection
dataset_03 https://zenodo.org/record/6098306/files/HiFi_synthetic_50x_03.fasta?download=1 fasta HiFi HiFi_collection
From Rules menu select Add / Modify Column Definitions
Click Add Definition button and select List Identifier(s): column A
Click Add Definition button and select URL: column B
Click Add Definition button and select Type: column C
Click Add Definition button and select Group Tag: column D
Click Add Definition button and select Collection Name: column E
Click Apply and press Upload
If working on a genome other than the example yeast genome, you can upload the VGP data from the VGP/Genome Ark AWS S3 bucket as follows:
Hands-on: Import data from GenomeArk
Open the file galaxy-uploadupload menu
Click on Choose remote files tab
Click on the Genome Ark button and then click on species
You can find the VGP data following this path: /species/${Genus}_${species}/${specimen_code}/genomic_data. Inside a given datatype directory (e.g.pacbio), select all the relevant files individually until all the desired files are highlighted and click the Ok button. Note that there may be multiple pages of files listed. Also note that you may not want every file listed.
Click on Operations on multiple datasets (check box icon) at the top of the history panel
Check all the datasets in your history you would like to include
Click For all selected.. and choose Build dataset list
Enter a name for your collection
Click Create List to build your collection
Click on the checkmark icon at the top of your history again
Import workflows from WorkflowHub
Once we have imported the datasets, the next step is to import the VGP workflows from the WorkflowHub server. WorkflowHub is a workflow management system which allows workflows to be FAIR (Findable, Accessible, Interoperable, and Reusable), citable, have managed metadata profiles, and be openly available for review and analytics.
Hands-on: Import a workflow
Click on the Workflow menu, located in the top bar.
Click on the Import button, located in the right corner.
In the section Import a Workflow from Configured GA4GH Tool Registry Servers (e.g. Dockstore), click on Search form.
In the TRS Server: workflowhub.eu menu you should type name:vgp
Click on the desired workflow, and finally select the latest available version.
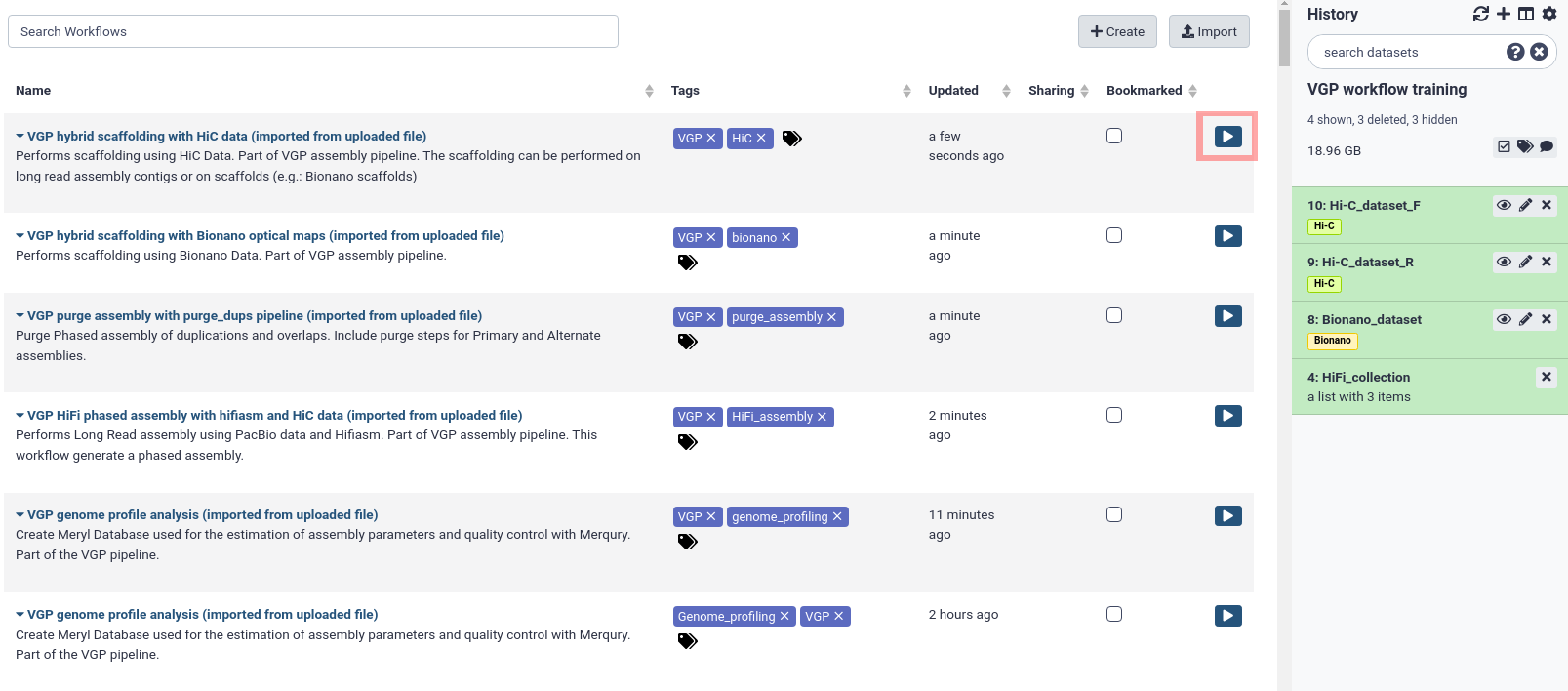
After that, the imported workflows will appear in the main workflow menu. In order to initialize the workflow, we just need to click in the workflow-runRun workflow icon, marked with a red square in figure 2.
Figure 2:Workflow main menu. The workflow menu lists all the workflows that have been imported. It provides useful information for organizing the workflows, such as last update and the tags. The worklows can be run by clicking in the play icon, marked in red in the image.
Once we have imported the datasets and the workflows, we can start with the genome assembly.
Comment: Workflow-centric Research Objects
In WorkflowHub, workflows are packaged, registered, downloaded and exchanged as Research Objects using the RO-Crate specification, with test and example data, managed metadata profiles, citations and more.
Now that our data and workflows are imported, we can run our first workflow. Before the assembly can be run, we need to collect metrics on the properties of the genome under consideration, such as the expected genome size according to our data. The present pipeline uses Meryl for generating the k-mer database and Genomescope2 for determining genome characteristics based on a k-mer analysis.
Hands-on: VGP genome profile analysis workflow
Click in the Workflow menu, located in the top bar
Click in the workflow-runRun workflow buttom corresponding to VGP genome profile analysis
In the Workflow: VGP genome profile analysis menu:
param-collection “Collection of Pacbio Data”: 7: HiFi_collection
“K-mer length”: 31
“Ploidy”: 2
Click on the Run workflow buttom
Comment: K-mer length
In this tutorial, we are using a k-mer length of 31. This can vary, but the VGP pipeline tends to use a k-mer length of 21, which tends to work well for most mammalian-size genomes. There is more discussion about k-mer length trade-offs in the extended VGP pipeline tutorial.
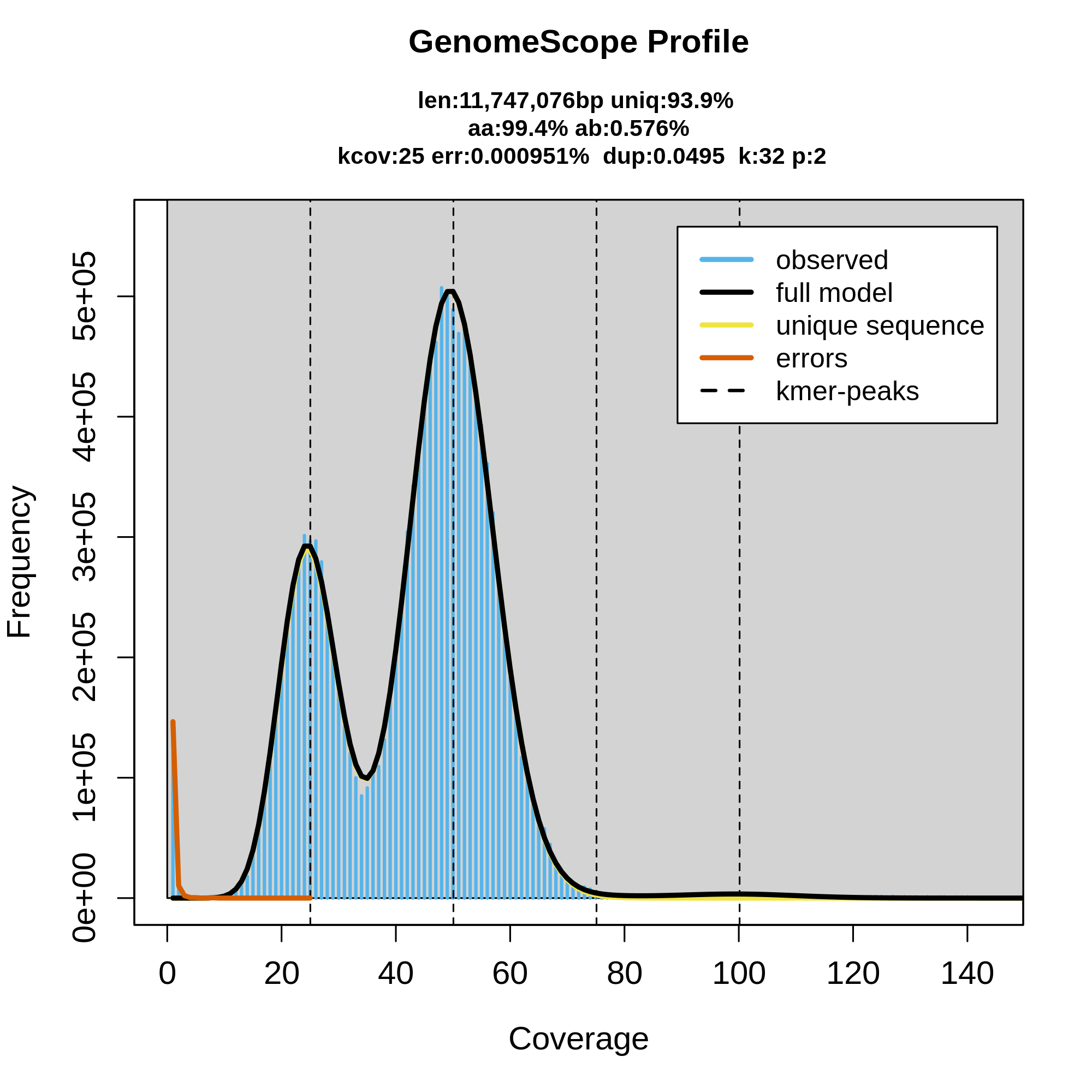
Once the workflow has finished, we can evaluate the linear plot generated by Genomescope (fig. 3), which includes valuable information such as the observed k-mer profile, fitted models and estimated parameters. This file corresponds to the dataset 26.
Figure 3: GenomeScope2 k-mer profile. The first peak located at about 25x corresponds to the heterozygous peak. The second peak at 50x, corresponds to the homozygous peak. The plot also includes information about the the inferred total genome length (len), genome unique length percent (uniq), overall heterozygosity rate (ab), mean k-mer coverage for heterozygous bases (kcov), read error rate (err), average rate of read duplications (dup) and k-mer size (k).
This distribution is the result of the Poisson process underlying the generation of sequencing reads. As we can see, the k-mer profile follows a bimodal distribution, indicative of a diploid genome. The distribution is consistent with the theoretical diploid model (model fit > 93%). Low frequency k-mers are the result of sequencing errors, and are indicated by the red line. GenomeScope2 estimated a haploid genome size of around 11.7 Mbp, a value reasonably close to the Saccharomyces genome size.
After genome profiling, the next step is to run the VGP HiFi phased assembly with hifiasm and HiC data workflow. This workflow uses hifiasm (HiC mode) to generate HiC-phased haplotypes (hap1 and hap2). This is in contrast to its default mode, which generates primary and alternate pseudohaplotype assemblies. This workflow includes three tools for evaluating assembly quality: QUAST, BUSCO and Merqury.
Hands-on: VGP HiFi phased assembly with hifiasm and HiC data workflow
Click in the Workflow menu, located in the top bar
Click in the workflow-runRun workflow buttom corresponding to VGP HiFi phased assembly with hifiasm and HiC data
In the Workflow: VGP HiFi phased assembly with hifiasm and HiC data menu:
param-file “Meryl database”: 12: Meryl on data 11, data 10, data 9: read-db.meryldb
param-file “HiC forward reads”: 3: Hi-C_dataset_F
param-file “HiC reverse reads”: 2: Hi-C_dataset_R
param-file “Genomescope summary dataset”: 19: Genomescope on data 13 Summary
“K-mer length”: 31
“Ploidy”: 2
“Is genome large (>100Mb)?”: No
Click on the Run workflow button
Comment: Input option order
Note that the order of the inputs may differ slightly.
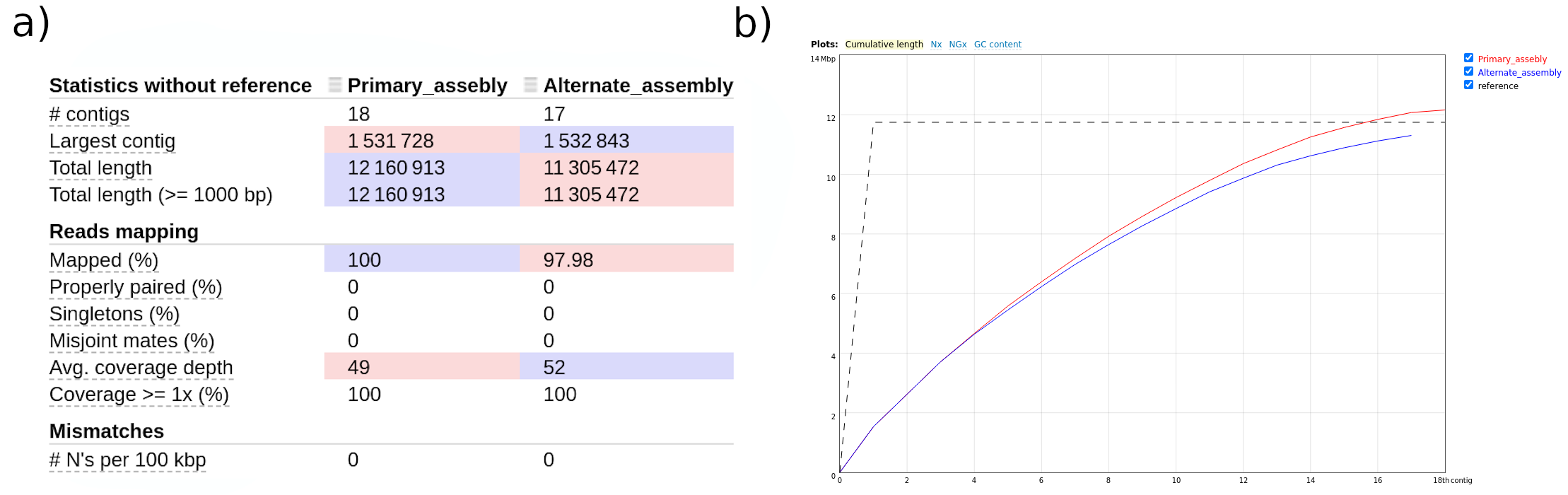
Let’s have a look at the HTML report generated by QUAST (fig. 4), which corresponds with the dataset 52. It summarizes some main assembly statistics, such as contig number, N50, assembly length, etc.
Figure 4: QUAST report. Statistics of the primary and alternate assembly (a). Cumulative length plot (b).
According to the report, both assemblies are quite similar; the primary assembly includes 18 contigs, whose cumulative length is around 12.2Mbp. The alternate assembly includes 17 contigs, whose total length is 11.3Mbp. As we can see in figure 4a, both assemblies come close to the estimated genome size, which is as expected since we used hifiasm-HiC mode to generate phased assemblies which lowers the chance of false duplications that can inflate assembly size.
Comment: Are you working with pri/alt assemblies?
This tutorial uses the hifiasm-HiC workflow, which generates phased hap1 and hap2 assemblies. The phasing helps lower the chance of false duplications, since the phasing information helps the assembler know which genomic variation is heterozygosity at the same locus versus being two different loci entirely. If you are working with primary/alternate assemblies (especially if there is no internal purging in the initial assembly), you can expect higher false duplication rates than we observe here with the yeast HiC hap1/hap2.
Question
What is the longest contig in the primary assembly? And in the alternate one?
What is the N50 of the primary assembly?
Which percentage of reads mapped to each assembly?
The longest contig in the primary assembly is 1.532.843 bp, and 1.532.843 bp in the alternate assembly.
The N50 of the primary assembly is 922.430 bp.
According to the report, 100% of reads mapped to both the primary assembly and the alternate assembly.
Next, we are going to evaluate the outputs generated by BUSCO. This tool provides quantitative assessment of the completeness of a genome assembly in terms of expected gene content. It relies on the analysis of genes that should be present only once in a complete assembly or gene set, while allowing for rare gene duplications or losses (Simão et al. 2015).
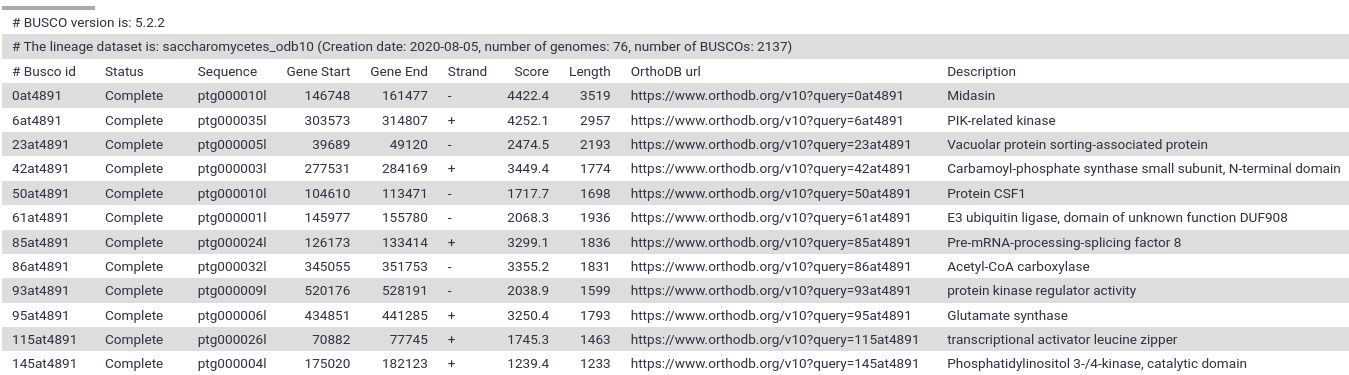
Figure 5: BUSCO full table. It contains the complete results in a tabular format with scores and lengths of BUSCO matches, and coordinates.
As we can see in the report, the results are simplified into four categories: complete and single-copy, complete and duplicated, fragmented and missing.
Question
How many complete BUSCO genes have been identified in the primary assembly?
How many BUSCOs genes are absent?
According to the report, our assembly contains the complete sequence of 2080 complete BUSCO genes (97.3%).
19 BUSCO genes are missing.
Despite BUSCO being robust for species that have been widely studied, it can be inaccurate when the newly assembled genome belongs to a taxonomic group that is not well represented in OrthoDB. Merqury provides a complementary approach for assessing genome assembly quality metrics in a reference-free manner via k-mer copy number analysis. Specifically, it takes our hap1 as the first genome assembly, hap2 as the second genome assembly, and the merylDB generated previously for k-mer counts. Like the other QC metrics we have been looking at, the VGP Hifiasm-HiC workflow will automatically generate the Merqury analysis.
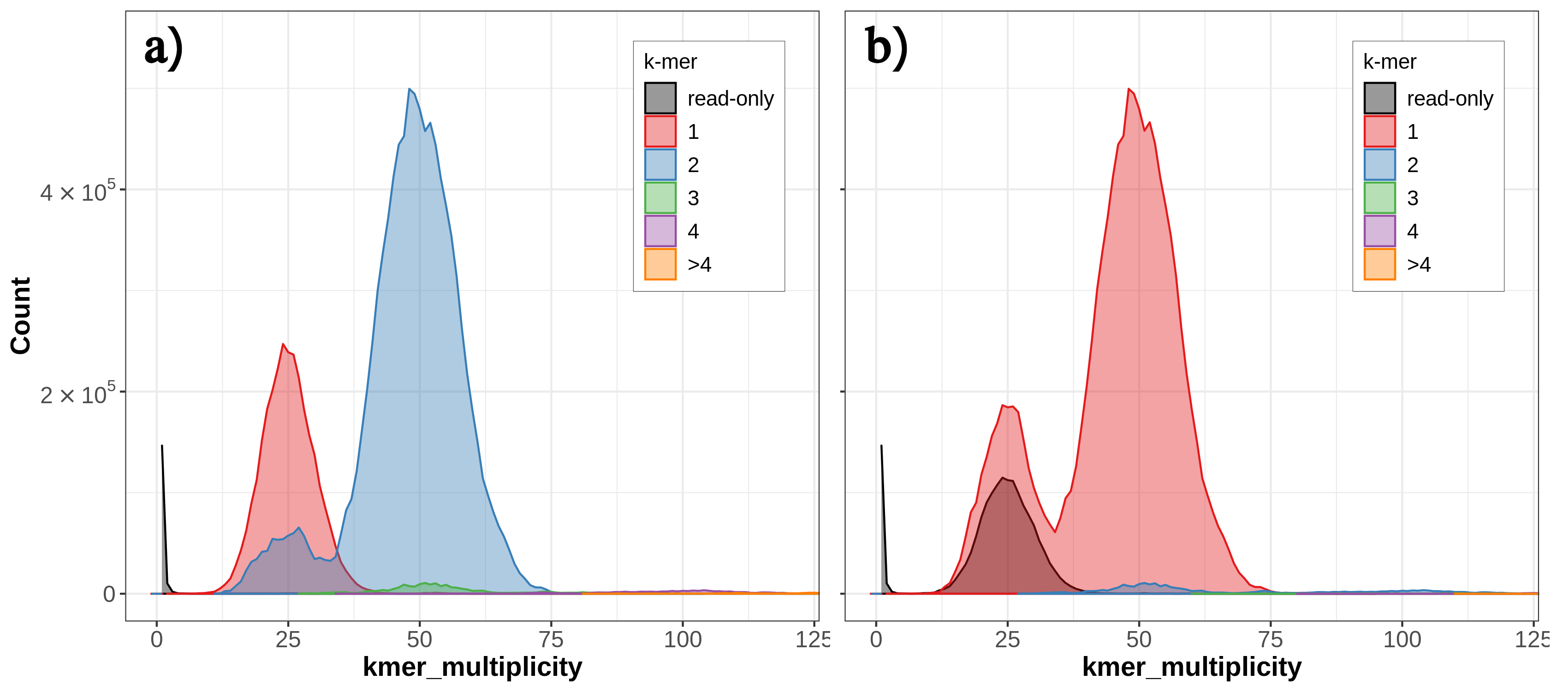
By default, Merqury generates three collections as output: stats, plots and QV stats. The “stats” collection contains the completeness statistics, while the “QV stats” collection contains the quality value statistics. Let’s have a look at the copy number (CN) spectrum plot, known as the spectra-cn plot. The spectra-cn plot looks at both of your assemblies (here, your haplotypes) taken together (fig. 6a). We can see a small amount of false duplications here: at the 50 mark on the x-axis, there is a small amount of k-mers present at 3-copy across the two assemblies (the green bump).
Figure 6: Merqury CN plot for these yeast haplotypes. The plot tracks the multiplicity of each k-mer found in the read set and colors it by the number of times it is found in a given assembly. Merqury connects the midpoint of each histogram bin with a line, giving the illusion of a smooth curve. K-mer distribution of both haplotypes (a). K-mer distribution of an individual haplotype (b)
Thus, we know there is some false duplication (the 3-copy green bump) present as 2-copy in one of our assemblies, but we don’t know which one. We can look at the individual copy number spectrum for each haplotype in order to figure out which one contains the 2-copy k-mers (i.e., the false duplications). In the Merqury spectra-CN plot for hap2 we can see the small bump of 2-copy k-mers at around the 50 mark on the x-axis (fig. 6b).
Now that we know which haplotype contains the false duplications, we can run the purging workflow to try to get rid of these duplicates.
An ideal haploid representation would consist of one allelic copy of all heterozygous regions in the two haplotypes, as well as all hemizygous regions from both haplotypes (Guan et al. 2019). However, in highly heterozygous genomes, assembly algorithms are frequently not able to identify the highly divergent allelic sequences as belonging to the same region, resulting in the assembly of those regions as separate contigs. In order to prevent potential issues in downstream analysis, we are going to run the VGP purge assembly with purge_dups workflow, which will allow to identify and reassign heterozygous contigs. This step is only necessary if haplotypic duplications are observed, and the output should be carefully checked for overpurging.
Hands-on: VGP purge assembly with purge_dups pipeline workflow
Click in the Workflow menu, located in the top bar
Click in the workflow-runRun workflow buttom corresponding to VGP purge assembly with purge_dups pipeline
In the Workflow: VGP purge assembly with purge_dups pipeline menu:
param-file “Hifiasm Primary assembly”: 39: Hifiasm HiC hap1
param-file “Hifiasm Alternate assembly”: 40: Hifiasm HiC hap2
param-file “Genomescope model parameters”: 20: Genomescope on data 13 Model parameters
Click in the Run workflow buttom
This workflow generates a large number of outputs, among which we should highlight the datasets 74 and 91, which correspond to the purged primary and alternative assemblies respectively.
Once the assemblies generated by hifiasm have been purged, the next step is to run the VGP hybrid scaffolding with Bionano optical maps workflow. It will integrate the information provided by optical maps with primary assembly sequences in order to detect and correct chimeric joins and misoriented contigs. In addition, this workflow includes some additonal steps for evaluating the outputs.
Hands-on: VGP hybrid scaffolding with Bionano optical maps workflow
Click in the Workflow menu, located in the top bar
Click in the workflow-runRun workflow buttom corresponding to VGP hybrid scaffolding with Bionano optical maps
In the Workflow: VGP hybrid scaffolding with Bionano optical maps menu:
param-file “Bionano data”: 1: Bionano_dataset
param-file “Hifiasm Purged Assembly”: 90: Purge overlaps on data 88 and data 33: get_seqs purged sequences (note: the data numbers may differ in your workflow, but it should still be tagged p1 from the purge_dups workflow)
Once the workfow has finished, let’s have a look at the assembly reports.
As we can observe in the cumulative plot of the file 119 (fig. 7a), the total length of the assembly (12.160.926 bp) is slightly larger than the expected genome size. With respect to the NG50 statistic (fig. 7b), the value is 922.430 bp, which is significantly higher than the value obtained during the first evaluation stage (813.039 bp). This increase in NG50 means this scaffolded assembly is more contiguous compared to the non-scaffolded contigs.
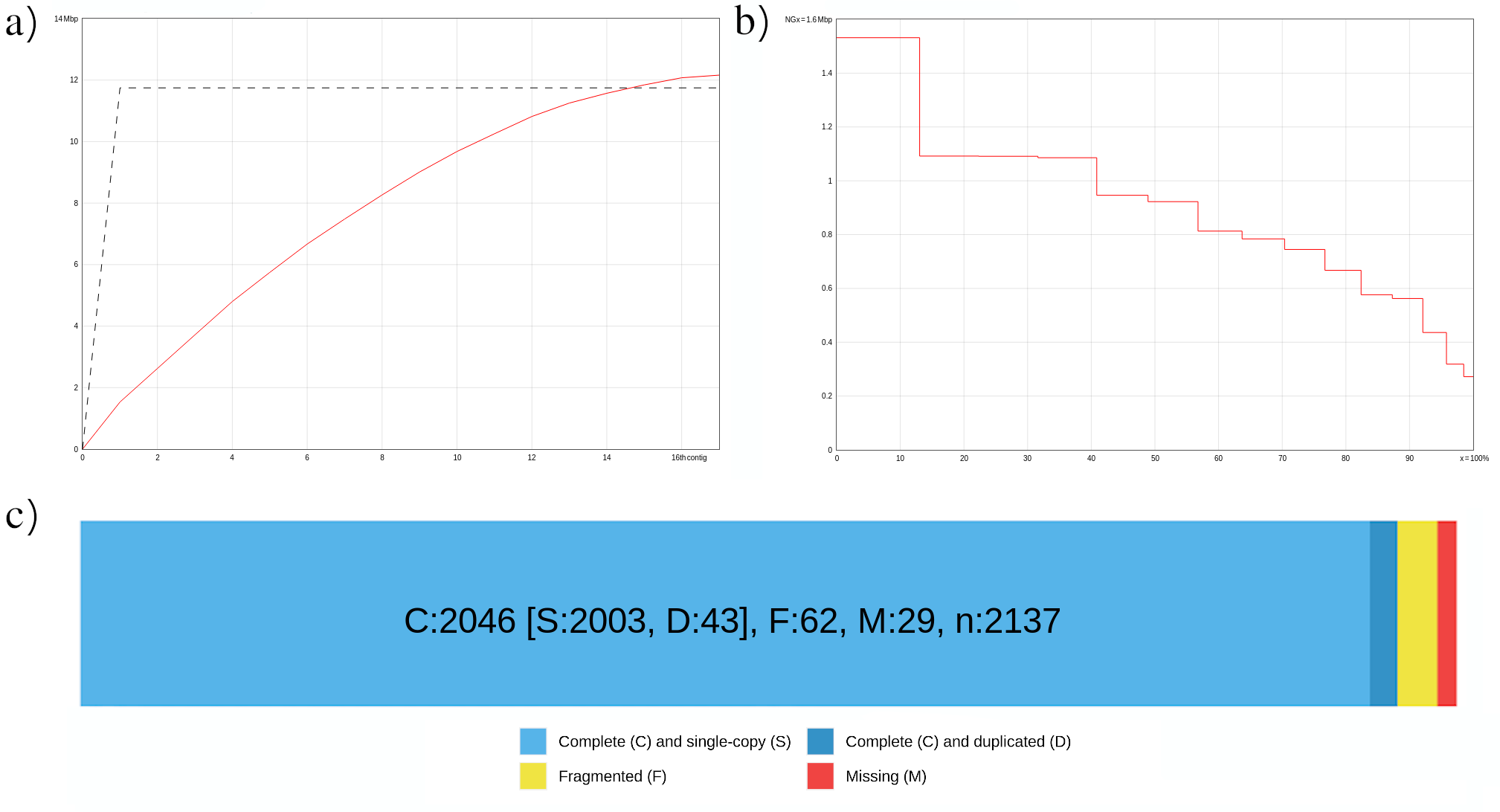
Figure 7: . Cumulative length plot (a). NGx plot. The y-axis represents the NGx values in Mbp, and the x-axis is the percentage of the genome (b). Assembly evaluation after runnig Bionano. BUSCO genes are defined as "Complete (C) and single copy (S)" when found once in the single-copy ortholog database, "Complete (C) and duplicated (D)" when found more than once, "Fragmented (F)" when genes are matching just partially, and "Missing (M)" when genes were expected but not detected (c).
It is also recommended to examine BUSCO outputs. In the summary image (fig. 7c), which can be found in the daset 117, we can appreciate that most of the universal single-copy orthologs are present in our assembly at the expected single-copy.
Question
How many scaffolds are in the primary assembly after the hybrid scaffolding?
What is the size of the largest scaffold? Has this changed with respect to the previous evaluation?
What is the percentage of completeness on the core set genes in BUSCO? Has Bionano scaffolding increased the completeness?
The number of contigs is 17.
The largest contig is 1.531.728 bp long. This value hasn’t changed. This is expected, as the VGP pipeline implementation of Bionano scaffolding does not allow for breaking contigs.
The percentage of complete BUSCOs is 95.7%. Yes, it has increased, since in the previous evaluation the completeness percentage was 88.7%.
In this final stage, we will run the VGP hybrid scaffolding with HiC data, which exploits the fact that the contact frequency between a pair of loci strongly correlates with the one-dimensional distance between them. This information allows further scaffolding the Bionano scaffolds using SALSA2, usually generating chromosome-level scaffolds.
Hands-on: VGP hybrid scaffolding with HiC data
Click in the Workflow menu, located in the top bar
Click in the workflow-runRun workflow buttom corresponding to VGP hybrid scaffolding with HiC data
In the Workflow: VGP hybrid scaffolding with HiC data menu:
param-file “Scaffolded Assembly”: 114: Concatenate datasets on data 110 and data 109
In order to evaluate the Hi-C hybrid scaffolding, we are going to compare the contact maps before and after running the HiC hybrid scaffolding workflow (fig. 8), corresponding to the datasets 130 and 141 respectively.
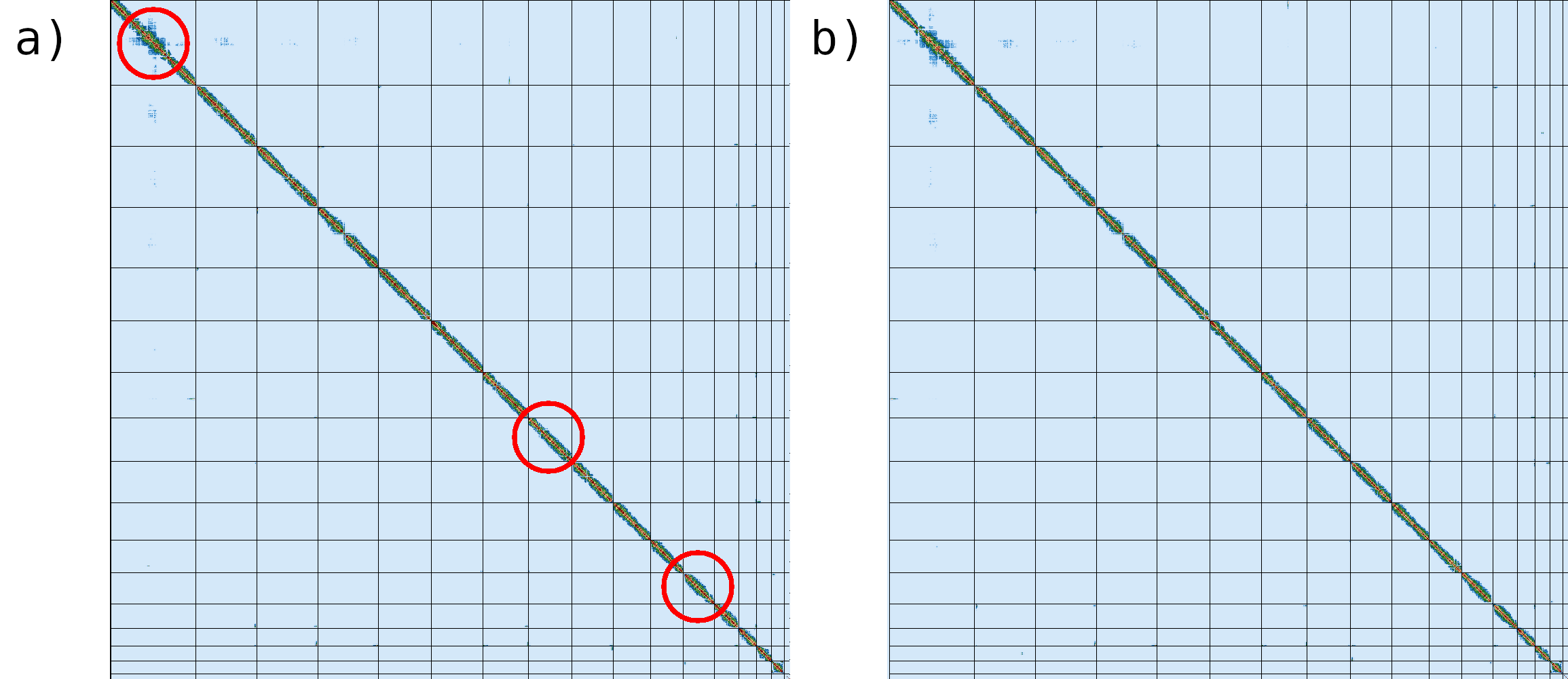
Figure 8: Hi-C maps generated by Pretext using Hi-C data. The red circles indicate the differences between the contact maps generated after (a) and before (b) Hi-C hybrid scaffolding.
The regions marked with red circles highlight the most notable difference between the two contact maps, where inversion has been fixed.
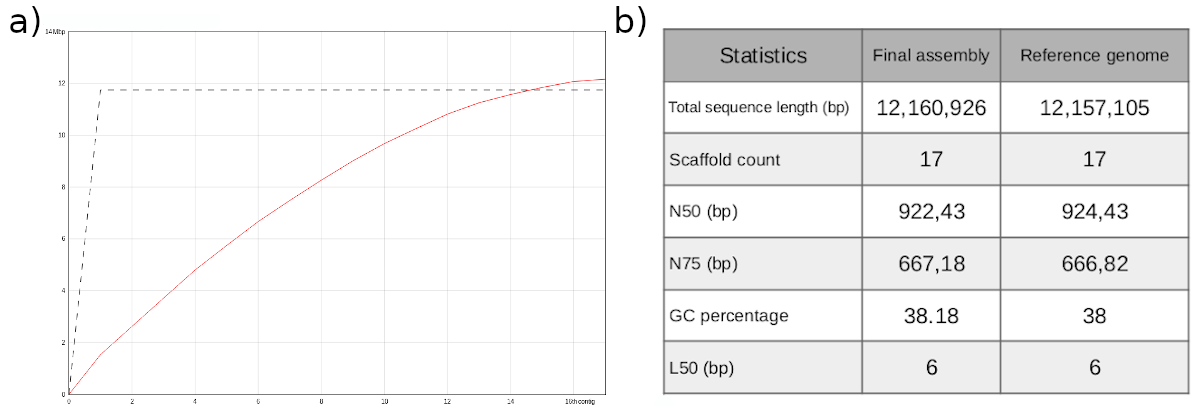
Figure 9: Comparison between the final assembly generated in this training and the reference genome. Contiguity plot using the reference genome size (a). Assemby statistics (b).
With respect to the total sequence length, we can conclude that the size of our genome assembly is almost identical to the reference genome (fig.9a,b). It is noteworthy that the reference genome consists of 17 sequences, while our assembly includes only 16 chromosomes. This is due to the fact that the reference genome also includes the sequence of the mitochondrial DNA, which consists of 85,779 bp. The remaining statistics exhibit very similar values (fig. 9b).
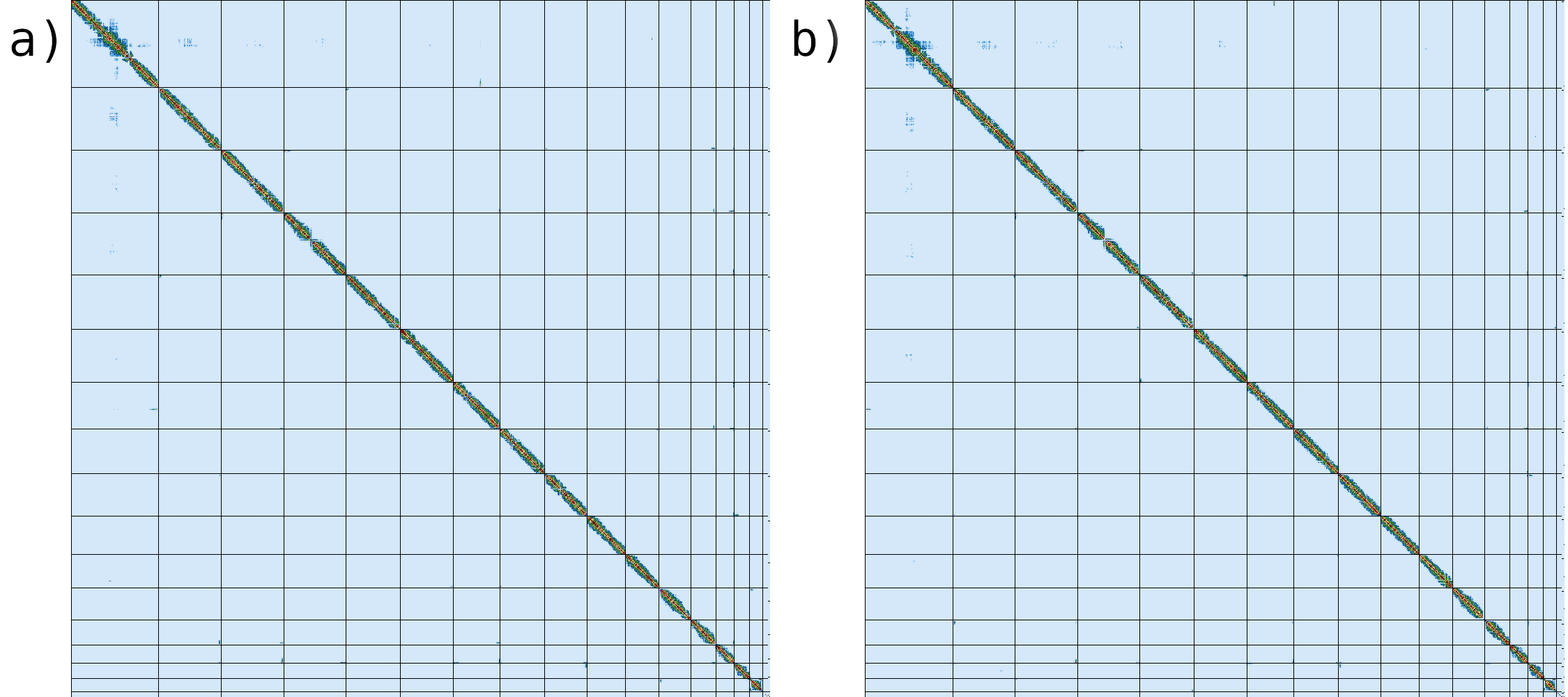
Figure 10: Comparison between contact maps generated using the final assembly (a) and the reference genome (b).
If we compare the contact map of our assembled genome (fig. 10a) with the reference assembly (fig. 10b), we can see that the two are indistinguishable, suggesting that we have generated a chromosome level genome assembly.
Key points
The VGP pipeline allows to generate error-free, near gapless reference-quality genome assemblies
The assembly can be divided in four main stages: genome profile analysis, HiFi long read phased assembly with hifiasm, Bionano hybrid scaffolding and Hi-C hybrid scaffolding
Simão, F. A., R. M. Waterhouse, P. Ioannidis, E. V. Kriventseva, and E. M. Zdobnov, 2015 BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31: 3210–3212. 10.1093/bioinformatics/btv351
Guan, D., S. A. McCarthy, J. Wood, K. Howe, Y. Wang et al., 2019 Identifying and removing haplotypic duplication in primary genome assemblies. 10.1101/729962
Rhie, A., S. A. McCarthy, O. Fedrigo, J. Damas, G. Formenti et al., 2021 Towards complete and error-free genome assemblies of all vertebrate species. Nature 592: 737–746. 10.1038/s41586-021-03451-0
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.
Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{assembly-vgp_workflow_training,
author = "Delphine Lariviere and Alex Ostrovsky and Cristóbal Gallardo and Brandon Pickett and Linelle Abueg",
title = "VGP assembly pipeline - short version (Galaxy Training Materials)",
year = "2022",
month = "10",
day = "18"
url = "\url{https://training.galaxyproject.org/training-material/topics/assembly/tutorials/vgp_workflow_training/tutorial.html}",
note = "[Online; accessed TODAY]"
}
@article{Batut_2018,
doi = {10.1016/j.cels.2018.05.012},
url = {https://doi.org/10.1016%2Fj.cels.2018.05.012},
year = 2018,
month = {jun},
publisher = {Elsevier {BV}},
volume = {6},
number = {6},
pages = {752--758.e1},
author = {B{\'{e}}r{\'{e}}nice Batut and Saskia Hiltemann and Andrea Bagnacani and Dannon Baker and Vivek Bhardwaj and Clemens Blank and Anthony Bretaudeau and Loraine Brillet-Gu{\'{e}}guen and Martin {\v{C}}ech and John Chilton and Dave Clements and Olivia Doppelt-Azeroual and Anika Erxleben and Mallory Ann Freeberg and Simon Gladman and Youri Hoogstrate and Hans-Rudolf Hotz and Torsten Houwaart and Pratik Jagtap and Delphine Larivi{\`{e}}re and Gildas Le Corguill{\'{e}} and Thomas Manke and Fabien Mareuil and Fidel Ram{\'{\i}}rez and Devon Ryan and Florian Christoph Sigloch and Nicola Soranzo and Joachim Wolff and Pavankumar Videm and Markus Wolfien and Aisanjiang Wubuli and Dilmurat Yusuf and James Taylor and Rolf Backofen and Anton Nekrutenko and Björn Grüning},
title = {Community-Driven Data Analysis Training for Biology},
journal = {Cell Systems}
}
Congratulations on successfully completing this tutorial!
 Delphine Lariviere
Delphine Lariviere Alex Ostrovsky
Alex Ostrovsky Cristóbal Gallardo
Cristóbal Gallardo Brandon Pickett
Brandon Pickett Linelle Abueg
Linelle Abueg Questions:
Questions: